|
Corresponding author: Іryna Drapak ( iradrapak@ukr.net ) Academic editor: Maya Georgieva
© 2019 Іryna Drapak, Borys Zimenkovsky, Lina Perekhoda, Hanna Yeromina, Kateryna Lipakova, Inna Demchuk, Marina Rakhimova.
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Drapak І, Zimenkovsky B, Perekhoda L, Yeromina H, Lipakova K, Demchuk I, Rakhimova M (2019) QSAR-analysis of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholine-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one’s derivatives as potential antioxidants. Pharmacia 66(1): 33-40. https://doi.org/10.3897/pharmacia.66.e35083
|
Abstract
Aim. The aim of study was to determine of the parameters of the molecular structure of new 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholine-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives and QSAR-analysis. The latter can be considered as the theoretical basis for de novo design of new potential antioxidants.
Materials and methods. 14 new derivatives of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholine-4-yl] propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one were involved in the study and their antioxidant activities were evaluated. Hyper-Chem 7.59 and BuildQSAR software were used for calculation of molecular descriptors and building the QSAR-models.
Results. The calculation of number of molecular descriptors (electronic, steric, geometric, energy) was carried out for the tested compounds: 14 derivatives of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholine-4-yl] propyl) -2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one. For QSAR analysis, the compounds studied were divided into a training and test sample. The correlations between the antioxidant activity level and abovementioned molecular descriptors were shown in multivariate linear QSAR-model: Activity = ∑хіаі + bі, where xi – molecular descriptor. Based on the analysis of the obtained QSAR-models, it was found that antioxidant activity increases with decreasing of the area, molecular volume, lipophilicity, polarisation and increasing the magnitude of the dipole moment. The increase in the energy of the bonds, the energy of inter-nuclear interactions, the energy of the lower vacant molecular orbit and the reduction of the energy of hydration and energy of the higher vacant molecular orbitals also results in an increase in the antioxidant activity. The greatest effect of effective charges on atoms on the antioxidant activity was detected: the increase in the charge value on the morpholine cycle Oxygen and the decrease in the charge size on the Sulphur atom of the thiazole ring and the Oxygen atom of the acetyl group. QSAR models with better statistics were selected. QSAR models obtained are characterised by high predictive ability, determined both by internal and external validation and can be used for virtual screening of the antioxidant activity of substances of this class of compounds.
Conclusions. 1). The study of the structure–activity relationships for 1-[2-(R-phenylimino)-4-methyl-3-(3- [morpholine-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives were carried out. 2). QSAR analysis revealed the following: polarisation, dipole moment, lipophilicity, energy parameters as well as the size of the molecule and its branching possessed the most significant effect on antioxidant activity; the antioxidant activities of the compounds were increased with the increase in their hydrophilic and reductive properties; the molecules with small volume and surface area showed the higher level of antioxidant activity. 3). Obtained QSAR models are proposed for antioxidant activity prediction within the above-mentioned row of compounds and can be considered as a theoretical basis for de novo design of new potential antioxidants.
Keywords
antioxidant activity, molecular descriptors, 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholine-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives, QSAR analysis
Introduction
In silico study and theoretical research including QSAR (Quantitative Structure-Activity Relationship) analysis are of special interest and a basis for design and directed synthesis of new drug-like molecules. QSAR-analysis consists of the identification and quantitative description of structural parameters or physico-chemical properties (descriptors) of a molecule in order to reveal the effect of each of them on the biological activity of a substance. Obtained QSAR models provide the information on the structural features of the molecules and outline the main directions for further design and optimisation of active compounds (
Currently, the role of free radicals, damage of biologically important molecules and oxidative stress are discussed. Such pathological conditions and diseases, such as cancer, atherosclerosis, Parkinson’s disease, staining processes, various types of ischaemia, cataract, neurodegenerative, cardiovascular diseases and aging processes are mainly associated with free radical oxidation and are considered as oxidative-stress related diseases and processes. Thus, the search for new efficient antioxidant/antiradicals entities is important for the prevention and therapy of the above-mentioned oxidative-stress related diseases (
Thiazole derivatives are promising in the search for biologically active compounds, since the thiazole frame is a powerful biophore fragment for the rational design of drug-like molecules. Thiazole derivatives possess various types of biological activity: anticonvulsant (
The aim of the study was to determine the molecular structure parameters of the new 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholin-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives and building the QSAR models as a theoretical basis for de novo design of new thiazole-based antioxidants.
Materials and methods
Fourteen new derivatives of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholin-4-yl]propyl)-2,3-dihydro-1,3-thiazole-5-yl]ethane-1-one, with established antioxidant activity, were used. Target thiazole-based compounds II (Fig.

Calculation of molecular descriptors was carried out using Hyper-Chem 7.5 software (HyperCube, Inc.) (licence for HyperChem 7.5 software is available for Danylo Halytsky Lviv National Medical University): BuildQSAR softaware was used for QSAR-model building (
The antioxidant activity (AOA) of the tested compounds was evaluated in vitro at the initiation of free radical processes by modelling the artificial oxidative stress, using the emulsion with yolk lipoprotein as a substrate for oxidation. Butylated hydroxytoluene (BHT) and quercetin (q) were taken as reference substances. The experiment was carried out under simulated conditions; the experiment variants included a control substance (DMSO solvent), solutions of reference substances (BHT, quercetin) and tested substances with a concentration in an incubation medium 0.3 kg/m3. Levels of compound activities were presented as a percentage of inhibition of the TBARs formation (tiobarbituric reactive substances) compared to BHT and quercetin activities: AOABHT (percentage of inhibition under BHT action) and AOAQ (percentage of inhibition under quercetin action) (
Antioxidant activity of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholin-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives (as inhibition % of the TBA-active products formation).
| № | АОАBHT | АОАQ |
|---|---|---|
| I | 45.78 | 58.25 |
| II a | 42.66 | 54.26 |
| II b | 41.09 | 52.28 |
| II c | 36.96 | 47.03 |
| II d | 5.81 | 7.39 |
| II e | 15.07 | 27.79 |
| II f | 16.08 | 20.26 |
| II g | 20.34 | 25.87 |
| II h | 32.06 | 40.79 |
| II i | 31.75 | 40.40 |
| II j | 31.65 | 40.26 |
| II k | 44.88 | 57.10 |
| II l | 44.72 | 56.90 |
| II m | 23.61 | 30.03 |
Results and discussion
The preliminary optimisation of the molecular structure of the investigated compounds was carried out using the molecular mechanics method MM+ (HyperChem software package) to achieve the RMS gradient value less than 0.1 kcal/(mol ∙ Å). The final minimisation of the energies of the investigated structures was carried out using AM1 semi-empirical quantum chemical method to achieve a RMS gradient value less than 0.01 kcal/(mol ∙ Å). The use of the AM1 method was due to the fact that it allowed the most accurate calculation of the electron-spatial structure of heterocyclic compounds containing Oxygen and Nitrogen atoms (
Molecular descriptors of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholin-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives.
| № | Ch_S | Ch_O(m) | Ch_N(m) | Ch_O | logP | R | P | D | S | V |
|---|---|---|---|---|---|---|---|---|---|---|
| I | -0.291 | -0.269 | -0.256 | 0 | 0.82 | 87.77 | 32.28 | 3.693 | 519.38 | 861.48 |
| II a | 0.467 | -0.266 | -0.250 | -0.274 | 0.68 | 110.2 | 39.91 | 5.052 | 584.10 | 1025.57 |
| II b | 0.461 | -0.261 | -0.250 | -0.272 | 0.83 | 114.49 | 41.74 | 4.874 | 604.44 | 1068.10 |
| II c | 0.461 | -0.261 | -0.251 | -0.274 | 0.99 | 118.77 | 43.58 | 5.036 | 628.34 | 1113.10 |
| II d | 0.458 | -0.267 | -0.250 | -0.270 | 0.99 | 118.77 | 43.58 | 4.930 | 632.07 | 1121.94 |
| II e | 0.514 | -0.268 | -0.258 | -0.284 | 0.99 | 118.77 | 43.58 | 2.245 | 611.81 | 1084.38 |
| II f | 0.467 | -0.267 | -0.248 | -0.278 | 0.99 | 118.77 | 43.58 | 5.412 | 631.99 | 1121.20 |
| II g | 0.461 | -0.261 | -0.250 | -0.271 | 0.99 | 118.77 | 43.58 | 4.966 | 642.83 | 1130.90 |
| II h | 0.700 | -0.265 | -0.262 | -0.380 | -0.31 | 116.58 | 42.38 | 12.871 | 607.52 | 1081.99 |
| II i | 0.464 | -0.267 | -0.250 | -0.269 | -0.31 | 116.58 | 42.38 | 5.422 | 628.10 | 1105.53 |
| II j | 0.466 | -0.268 | -0.259 | -0.279 | -0.31 | 116.58 | 42.38 | 5.662 | 627.92 | 1103.68 |
| II k | 0.482 | -0.268 | -0.261 | -0.280 | 0.46 | 114.92 | 41.84 | 5.429 | 602.54 | 1062.59 |
| II l | 0.478 | -0.268 | -0.259 | -0.278 | 0.46 | 114.92 | 41.84 | 5.018 | 608.96 | 1070.60 |
| II m | 0.476 | -0.266 | -0.251 | -0.274 | 0.46 | 114.92 | 41.84 | 4.812 | 606.84 | 1068.49 |
Energy parameters of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholin-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives.
| № | TE | BE | IAE | EE | CCI | HF | HOMO | LUMO | EH |
|---|---|---|---|---|---|---|---|---|---|
| I | -74183 | -3944 | -70239.1 | -495470 | 421286 | 7.538 | -8.270 | -0.149 | -7.78 |
| II a | -97589 | -5078 | -92510.6 | -774500 | 676911 | -4.85 | -8.111 | -0.192 | -3.57 |
| II b | -101183 | -5361 | -95822.3 | -831881 | 730698 | -11.88 | -8.070 | -0.170 | -2.57 |
| II c | -104776 | -5642 | -99134.1 | -885438 | 780661 | -17.87 | -8.050 | -0.172 | -1.65 |
| II d | -104778 | -5643 | -99134.1 | -881757 | 776978 | -19.49 | -8.039 | -0.128 | -1.49 |
| II e | -104732 | -5597 | -99134.1 | -910580 | 805848 | 26.48 | -6.894 | -0.848 | -1.70 |
| II f | -104777 | -5643 | -99134.1 | -876830 | 772052 | -19.12 | -8.058 | -0.182 | -1.53 |
| II g | -104778 | -5644 | -99134.1 | -876880 | 772102 | -19.93 | -8.084 | -0.116 | -1.45 |
| II h | -108497 | -5384 | -103112 | -885766 | 777269 | 23.99 | -7.034 | -1.457 | -2.85 |
| II i | -108563 | -5451 | -103112 | -878934 | 770370 | -42.60 | -8.110 | -0.116 | -4.94 |
| II j | -108563 | -5450 | -103112 | -877278 | 768714 | -42.07 | -8.054 | -0.247 | -5.23 |
| II k | -105893 | -5061 | -100831 | -833546 | 727653 | -10.88 | -8.173 | -0.343 | -3.15 |
| II l | -105893 | -5062 | -100831 | -827008 | 721114 | -11.48 | -8.232 | -0.364 | -3.22 |
| II m | -105894 | -5062 | -100831 | -824380 | 718485 | -12.07 | -8.215 | -0.319 | -3.25 |
Based on the obtained results of charge values, the following conclusions can be made. Effective charges on Sulphur atoms of the thiazole cycle have additional values: least value of 0.458 (compound IId) and largest value of 0.700 (compound IIh), the charge on the Sulphur atom in the unsubstituted thiourea has an electronegative value of (-)0.291. The effective charges on the Oxygen atom for the test compounds take values from (-) 0.270 (IId) to (-) 0.370 (IIh). Thus, amongst the studied compounds, the most electronegative is the Oxygen atom and the most positive charge is on the Sulphur atom in the IIh compound, which contains the methoxy group in position 2 of the phenyl fragment. The compound IId, with methyl substituents in the phenyl fragment at positions 2,6, is characterised by the lowest value of the charge on the Sulphur atom and the lowest value on the Oxygen atom. The effective charges on the Oxygen and Nitrogen atoms of the morpholine cycle for the studied compounds are characterised by almost identical values of (-) 0.26 (Ch_O (m)), (-) 0.25 Ch_N (m).
Comparing the values of refractivity and polarisability, which are known to characterise the mobility of the molecule electron shell (
The reactivity of a substance depends on the hydrophilic-hydrophobic balance of the molecule, the quantitative criterion of which is the value of the dipole moment. The value of the dipole moment of the investigated compounds has very different values from 2.245 (IIe) to 12.871 (IIh).
An important characteristic of substances is lipophilicity (logP). It is known that an increase of lipophilicity leads to an increase in the penetration of biologically active substances through cell membranes, but a reduction in the water solubility and elimination from the body (
The surface area of the molecule and the volume of the molecule were the smallest for compound I and the largest value of these parameters were for compound IIg.
Based on the energy parameters, it can be concluded that compound I has the minimum values for total energy of the molecule, binding energies, energy of isolated atoms, electronic energy and energy of internuclear interactions. The energies of the frontier orbitals are responsible for the donor-acceptor properties of the molecules: the minimum HOMO value is in compound I (-8.270) and the largest value in compound IIe (-6.894); the least LUMO value is in IIh compound (-1.457), the largest value in IIg and IIi compounds (-0.116). The calculated energy gap (the energy difference between the lower occupied and the upper vacant molecular orbitals) value of the molecules for the studied compounds is within the range of 7.8–7.9, with the exception I compound having the value of 8.121, IIe compound –(6.046 value) and IIh compound - (5.577 value).
The structure-activity relationships were studied using the calculated descriptors and antioxidant activity values for the studied compounds. For this purpose, the construction of mathematical QSAR models was carried out using the BuildQSAR programme and the GA-MLRA method, which had allowed the generation of one- or multi-parameter models with a maximum value of the correlation coefficient (r) and the minimum value of standard deviation (s). Obtained QSAR models were analysed by the Fisher coefficient (F) value and “leave-one-out” method under confirmation of the predictive model capability, which had been verified by the value of the cross-validation coefficient (Q2), calculated using the sum of squares of prediction error (SRRESS) (
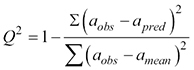 ,
,
where aobs is the observed or experimental activity, apred is the activity predicted by a certain model, amean is the average activity.
The QSAR models were presented in the next formula:
% = a + b ∙ X1 + c ∙ X2 + d ∙ X3,
where the activity parameter % is AOABHT or AOAQ and X1, X2, X3 are molecular descriptors.
The mathematical dependence between the inhibition of BHT and quercetin was described by the equation: AOABHT = +0.784 AOA_Q +0.0598. Therefore, the parameter of activity AOABHT was used in QSAR-analysis.
The total sample of compounds was divided into three groups according to the values of their antioxidant activity: 1 group - active compounds (AOABHT more than 40%): І, IIа, IIb, IIk, IIl; 2 group - average activity AOABHT 25–40%: IIc, IIh, IІi, IIj and group 3 - compounds with a low activity AOABHT of less than 25%: IId, IIe, IIf, IIg, IIm. For QSAR analysis, the compounds studied were divided into a training and test sample. The test sample includes compounds IIb, IIe, IIh, IIl. The remaining compounds were a training sample. Thus, the ratio of compounds in the training and test sample was 78.57% and 21.43%. Compound І was also in the training and in the test sample.
When one-parameter QSAR-models were built in the training sample, the largest correlation coefficient r = 0.717 was observed with the use of the descriptor – the area of the molecule. The anti-oxidant activity increases with the decrease of the area of the molecule:
1. AOABHT = -0.224 (±0.174) S +153.343(±96.297)
(n=010; r=0.717; s=9.857; F=8.446; Q2=0.126; SPRESS=13.212)
The QSAR model is characterised by low prognostic ability.
From the two-parameter QSAR models in the training sample of compounds, model 2 was selected, which was characterised by a higher correlation coefficient (r) and predictive power (Q2):
2. AOABHT = -0.367(±0.213) S + 15.431(±16.563) D + 154.458(±81.478)
(n = 010; r = 0.841; s = 8.180; F = 8.440; Q2 = 0.514; SPRESS = 10.538)
When three-parameter QSAR-models were obtained in the training sample, the correlation coefficients had high values of 0.959–0.841.
3. AOABHT = -0.357(±0.106)V + 35.910(±13.811)D + 3546.238(±1827.550) Ch_O(m) + 1175.132(±526.812)
(n = 010; r = 0.959; s = 4.621; F = 22.950; Q2 = 0.809; SPRESS = 7.126)
4. AOABHT = -0.509(±0.189) S + 22.475(±13.016) D + 2498.261(±2004.255) Ch_O(m) + 861.58139 (±570.222154)
(n = 010; r = 0.938; s = 5.647; F = 14.705; Q2 = 0.240; SPRESS = 14.229)
5. AOABHT = + 0.051(±0.020) BE + 30.985(±17.148) D + 3696.943(±2519.472) Ch_O(m) + 1124.146(±699.651)
(n = 010; r = 0.926; s = 6.143; F = 12.117; Q2 = 0.517; SPRESS = 11.800)
6. AOABHT = -0.835(±0.494)V-0.00048(±0.00032)EE + 1911.515(±2428.216)Ch_O(m) + 1040.745 (±803.539)
(n = 010; r = 0.898; s = 7.176; F = 8.343; Q2 = 0.410; SPRESS = 19.381)
7. AOABHT = -0.880(±0.535)V + 0.00056(±0.00039)CCI + 1737.653(±2426.985)Ch_O(m) + 1035.634(±814.382)
(n = 010; r = 0.895; s = 7.275; F = 8.065; Q2 = 0.271; SPRESS = 18.401)
8. AOABHT = -7.938(±4.034)P + 33.855(±22.349)D + 3349.00072(±2943.253)Ch_O(m) + 1079.513(±836.794)
(n = 010; r = 0.893; s = 7.331; F = 7.912; Q2 = 0.227; SPRESS = 14.349)
9. AOABHT = -14.07748(±14.38322)logP-0.311(±0.177)S + 2497.805(±2977.171)Ch_O(m) + 873.634(±853.328)
(n = 010; r = 0.872; s = 7.979; F = 6.367; Q2 = 0.422; SPRESS = 12.412)
10. AOABHT = -0.632(±0.731)V + 0.00036(±0.00062)CCI + 8.011(±29.439)D + 407.662(±439.413)
(n = 010; r = 0.852; s = 8.553; F = 5.281; Q2 = 0.042; SPRESS = 16.661)
11. AOABHT = -0.903(±0.814)V + 0.00066(±0.00079)CCI-32.793(±119.469)Ch_S + 536.002(±371.253)
(n = 010; r = 0.852; s = 8.549; F = 5.289; Q2 = 0.452; SPRESS = 11.454)
12. 12. AOABHT = -0.713(±0.650)V -0.826(±5.486)EH + 0.00047(±0.00046)CCI + 456.532(±392.891)
(n = 010; r = 0.844; s = 8.752; F = 4.955; Q2 = 0.011; SPRESS = 16.230)
13. AOABHT = -0.814(±0.761)V + 0.00053(±0.00056)CCI + 14.946(±106.470)LUMO + 525.610(±430.731)
(n = 010; r = 0.844; s = 8.764; F = 4.936; Q2 = 0.047; SPRESS = 16.701)
14. AOABHT = -0.384(±0.275)S + 11.623(±102.755)LUMO + 16.777(±21.778)D + 158.873(±97.849)
(n = 010; r = 0.843; s = 8.783; F = 4.904; Q2 = 0.466; SPRESS = 11.921)
15. AOABHT = -1.499 (±17.015) logP -0.357(±0.262)S + 14.219(±22.873)D + 155.841(±91.292)
(n = 010; r = 0.842; s = 8.804; F = 4.872; Q2 = 0.478; SPRESS = 12.206)
16. AOABHT = -0.357(±0.299)S-8.698(±151.229)HOMO + 15.674(±18.799)D + 76.83871(±1352.516)
(n = 010; r = 0.841; s = 8.822; F = 4.844; Q2 = 0.350; SPRESS = 13.153)
17. AOABHT = -6.567(±15.048)logP -0.329(±0.298)S -61.493(±148.169)Ch_O + 199.959(±135.929)
(n = 010; r = 0.812; s = 9.531; F = 3.863; Q2 = 0.448; SPRESS = 16.069)
Based on the analysis of the obtained QSAR-models, it was found that antioxidant activity increases with decreasing of the area, molecular volume, lipophilicity, polarisation and increasing the magnitude of the dipole moment. The increase in the energy of the bonds, the energy of inter-nuclear interactions, the energy of the lower vacant molecular orbit and the reduction of the energy of hydration and energy of the higher vacant molecular orbitals also result in an increase in the antioxidant activity. The greatest effect of effective charges on atoms on the anti-oxidant activity was detected: the increase in the charge value on the morpholine cycle Oxygen and the decrease in the charge size on the Sulphur atom of the thiazole ring and the Oxygen atom of the acetyl group.
From the received QSAR models, models with the highest correlation coefficient and predictive power of 2, 3, 5, 15 were selected. To estimate the accuracy of the predictive ability of the received QSAR models, the prediction error values for compounds of the training sample were calculated (Table
Observed and predicted antioxidant activities and residual for QSAR- model 2, 3, 5, 15 for compounds of the training sample.
| № | QSAR- model 2 | QSAR- model 3 | ||||
|---|---|---|---|---|---|---|
| Observed antioxidant activity | Predicted antioxidant activity | Residual | Observed antioxidant activity | Predicted antioxidant activity | Residual | |
| I | 45.780 | 45.283 | 0.497 | 45.780 | 45.926 | -0.146 |
| II a | 42.660 | 45.078 | -2.418 | 42.660 | 46.722 | -4.062 |
| II c | 36.960 | 23.732 | 13.228 | 36.960 | 32.596 | 4.364 |
| II d | 5.810 | 17.796 | -11.986 | 5.810 | 4.353 | 1.457 |
| II f | 16.080 | 23.517 | -7.437 | 16.080 | 21.997 | -5.917 |
| II g | 20.340 | 13.628 | 6.712 | 20.340 | 23.712 | -3.381 |
| II i | 31.750 | 27.513 | 4.219 | 31.750 | 27.885 | 3.865 |
| II j | 31.650 | 33.054 | -1.040 | 31.650 | 33.619 | -1.969 |
| II k | 44.880 | 42.228 | 2.852 | 44.880 | 39.937 | 4.943 |
| II m | 23.610 | 27.675 | -4.065 | 23.610 | 22.764 | 0.846 |
| № | QSAR- model 5 | QSAR- model 15 | ||||
| I | 45.780 | 43.970 | 1.810 | 45.780 | 45.552 | 0.228 |
| II a | 42.660 | 39.588 | 3.072 | 42.660 | 44.496 | -1.836 |
| II c | 36.960 | 28.999 | 7.961 | 36.960 | 23.287 | 13.673 |
| II d | 5.810 | 3.450 | 2.360 | 5.810 | 17.598 | -11.788 |
| II f | 16.080 | 18.w404 | -2.324 | 16.080 | 22.783 | -6.703 |
| II g | 20.340 | 26.725 | -6.385 | 20.340 | 13.517 | 6.823 |
| II i | 31.750 | 28.459 | 3.291 | 31.750 | 28.628 | 3.122 |
| II j | 31.650 | 32.225 | -0.575 | 31.650 | 33.810 | -2.160 |
| II k | 44.880 | 44.742 | 0.138 | 44.880 | 41.758 | 3.122 |
| II m | 23.610 | 30.958 | -7.328 | 23.610 | 28.092 | -4.482 |
Figures


QSAR-model 3 is characterised by the best statistical indicators. Since in QSAR-models 2 and 15 for compounds IIc and IId, there was a large discrepancy between the observed and predicted anti-oxidant activities and these compounds were removed from the training sample for these models. The obtained QSAR models 2-1 and 15-1 were characterised by better statistical indicators.
2-1. AOABHT = -0.337(±0.169)S +13.190 (±12.625)D +148.870(±58.968)
(n=008; r=0.920; s=5.354; F=13.709; Q2=0.495; SPRESS=16.668)
15-1. AOABHT = -6.725(±7.142)logP-0.475 (±0.122)S +19.148(±9.752)D +194.955(±40.710)
(n=008; r=0.985; s=3.241; F=44.855; Q2=0.638; SPRESS=24.412)
Using the obtained QSAR models with the best statistical parameters, the activity of the compounds from the test sample was predicted. Table
Values of experimental antioxidant activity and activity predicted by QSAR models 2-1, 3, 5, and 15-1 for compounds from test sample.
| № | Experimental antioxidant activity | Predicted antioxidant activity for QSAR- models and residuals | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2-1 | Residual | 3 | Residual | 5 | Residual | 15-1 | Residual | ||
| І | 45.78 | 45.28 | 0.49 | 45.93 | -0.15 | 43.97 | 1.81 | 45.55 | 0.23 |
| IIb | 41.09 | 33.95 | 7.14 | 42.16 | -1.07 | 38.27 | 2.82 | 33.56 | 7.53 |
| IIe | 15.07 | 15.90 | -0.83 | 18.97 | -3.9 | 9.61 | 6.09 | 16.87 | -1.17 |
| IIh | 32.06 | 34.23 | -2.17 | 34.87 | -2.81 | 34.72 | -2.66 | 33.23 | -1.17 |
| IIl | 44.72 | 33.63 | 11.09 | 38.36 | 6.36 | 35.07 | 9.65 | 33.72 | 11 |
The predictive ability of the constructed models, tested with the use of compounds from the test sample, was quantified by the value of the cross-validation LGO coefficient Q2, which were for QSAR models 2-1, 3, 5, 15-1 – 0.530, 0.769, 0.608, 0.665, respectively. Thus, it can be assumed that the QSAR models obtained are characterised by high predictive ability, determined both by internal and external validation and can be used for virtual screening of the antioxidant activity of substances of this class of compounds.
Conclusions
- The study of the structure–activity relationships for 1-[2-(R-phenylimino)-4-methyl-3-(3- [morpholine-4-yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives was carried out.
- QSAR analysis revealed the following: polarisation, dipole moment, lipophilicity, energy parameters, as well as the size of the molecule and its branching, possessed the most significant effect on antioxidant activity; the antioxidant activity of the compounds was increased with the increasing of their hydrophilic and reductive properties; the molecules with small volume and surface area showed the higher level of antioxidant activity.
- Obtained QSAR-models are proposed for antioxidant activity prediction within the above-mentioned row of compounds and can be considered as a theoretical basis for de novo design of new potential antioxidants.
References
- Abdel-Wahab BF, Mohamed SF, Amr AE-GE, Abdalla MM (2008) Synthesis and reactions of thiosemicarbazides, triazoles, and Schiff bases as antihypertensive α-blocking agents. Monatshefte für Chemie – Chemical Monthly 139: 1083–1090. https://doi.org/10.1007/s00706-008-0896-2
- Andreani A, Leoni A, Locatelli A, Morigi R, Rambaldi M (2013) Chemopreventive and antioxidant activity of 6-substituted imidazo[2,1-b]thiazoles European Journal Medicinal Chemistry 68: 412–421. https://doi.org/10.1016/j.ejmech.2013.07.052
- De Olivera DB, Gaudio AC (2000) BuildQSAR: A new computer program for QSAR Studies. Quantitative Structure-Activity Relationships 19(6): 599–601. https://doi.org/10.1002/1521-3838(200012)19:6%3C599::AID-QSAR599%3E3.0.CO;2-B
- El-Sabbagh OI, Baraka MM, Ibrahim SM, Pannecouque C, Andrei G, Snoeck R, Balzarini J, Rashad AA (2009) Synthesis and antiviral activity of new pyrazole and thiazole derivatives. European Journal Medicinal Chemistry 44: 3746–3753. https://doi.org/10.1016/j.ejmech.2009.03.038
- Giri RS, Thaker HM, Giordano T, Williams J, Rogers D, Sudersanam V, Vasu KK (2009) Design, synthesis and characterization of novel 2-(2,4-disubstituted-thiazole-5-yl)-3-aryl-3H-quinazoline-4-one derivatives as inhibitors of NF-κB and AP1 mediated transcription activation and as potential anti-inflammatory agents. European Journal Medicinal Chemistry 44: 2184–2189. https://doi.org/10.1016/j.ejmech.2008.10.031
- Golbraikh A, Tropsha A (2002) Beware of q2. Journal of Molecular Grafics and Modelling 20(4): 269–276. https://doi.org/10.1016/S1093-3263(01)00123-1
- HyperCube (2019) Hyperchem Software. Hypercube, Inc, Gainesville. http://www.hyper.com
- Kumar V, Khan AA, Tripathi A, Dixit PK, Bajaj UK (2015) Role of oxidative stress in various diseases: Relevance of dietary antioxidants The Journal of Phytopharmacology 4(2): 126–132. http://www.phytopharmajournal.com/Vol4_Issue2_13.pdf
- Lavecchia A, Di Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Current Medicinal Chemistry 20(23): 2839–2860. https://doi.org/10.2174/09298673113209990001
- Lionta E, Spyrou G, Vassilatis DK, Cournia Z (2014) Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Current Topics in Medicinal Chemistry 14: 1923–1938. https://doi.org/10.2174/1568026614666140929124445
- Lipkowitz KB, Boyd DB (1990) Reviews in Computational Chemistry (Vol. 10). VCH Publishers, New York, 20 pp. https://onlinelibrary.wiley.com/doi/pdf/10.1002/9780470125878.fmatter
- Perekhoda L, Yeromina H, Drapak I, Kobzar N, Smolskiy O, Demchenko N (2017) The antioxidant properties of 1-[2-(R-phenylimino)-4-methyl-3-(3-[morpholine-4yl]propyl)-2,3-dihydro-1,3-thiazol-5-yl]ethane-1-one derivatives under conditions of artificial oxidative stress in vitro. Saudi Journal of Medical and Pharmaceutical Sciences 3(1): 55–59. https://pdfs.semanticscholar.org/213b/81ea6e156efeb587ba26d9a8d989e1872daf.pdf
- Satoh A, Nagatomi Y, Hirata Y, Ito S, Suzuki G, Kimura T, Maehara S, Hikichi H, Satow A, Hata M, Ohta H, Kawamoto H (2009) Discovery and in vitro and in vivo profiles of 4-fluoro-N-[4-[6-(isopropylamino)pyrimidin-4-yl]-1,3-thiazol-2-yl]-N-methylbenzamide as novel class of an orally active metabotropic glutamate receptor 1 (mGluR1) antagonist. Bioorganic & Medicinal Chemistry Letters 19: 5464–5468. https://doi.org/10.1016/j.bmcl.2009.07.097
- Szabo A, Ostlund NS (1989) Modern Quantum Chemistry. McGraw-Hill, New York, 466 pp. https://chemistlibrary.files.wordpress.com/2015/02/modern-quantum-chemistry.pdf
- Taha M, Ismail NH, Jamil W, Khan KM, Salar U, Kashif SM, Rahim F, Latif Y (2015) Synthesis and evaluation of unsymmetrical heterocyclic thioureas as potent β-glucuronidase inhibitors. Medicinal Chemistry Research 24(8): 3166–3173. https://doi.org/10.1007/s00044-015-1369-x
- Todeschini R, Consonni V (2000) Handbook of Molecular Descriptors. Wiley-VCH, Toronto, 667 pp. https://doi.org/10.1002/9783527613106
- Todeschini R, Consonni V (2009) Molecular Descriptors for Chemoinformatic (2 vols). WILEY-VCH, Weinheim, 967 pp. https://doi.org/10.1002/9783527628766
- Yeromina HO, Drapak IV, Perekhoda LO, Yaremenko VD, Demchenko AM (2016) Synthesis of 2-(4-aryl(adamantyl)-2-phenyliminothiazol-3-yl)-ethanol derivatives and prediction of their biological activity. Der Pharma Chemica 8(3): 64–70. http://www.scopus.com/inward/record.url?eid=2-s2.0-84962637541&partnerID=MN8TOARS