Research Article |
|
Corresponding author: Taufik Muhammad Fakih ( taufikmuhammadf@unisba.ac.id ) Academic editor: Emilio Mateev
© 2024 Taufik Muhammad Fakih, Ritmaleni, Rahadian Zainul, Muchtaridi Muchtaridi.
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Fakih TM, Ritmaleni, Zainul R, Muchtaridi M (2024) Molecular docking-based virtual screening and computational investigations of biomolecules (curcumin analogs) as potential lead inhibitors for SARS-CoV-2 papain-like protease. Pharmacia 71: 1-19. https://doi.org/10.3897/pharmacia.71.e123948
|
Abstract
In the effort to combat SARS-CoV-2 infection, researchers are currently exploring the repurposing of conventional antiviral drugs, despite their limited efficacy. The SARS-CoV-2 virus encodes a papain-like protease (PLpro), which not only plays a crucial role in viral replication but also cleaves ubiquitin and interferon-stimulated gene 15 protein (ISG15) from host proteins, making it a prime target for the development of new antiviral medications. In this study, we conducted a multi-step in silico screening to identify novel, noncovalent PLpro inhibitors. Curcumin, an antioxidant derived from turmeric rhizomes (Curcuma longa L.), has undergone extensive preclinical investigations and shown significant efficacy against viruses and other ailments in both laboratory and animal studies. However, the pharmacological limitations of curcumin have prompted the synthesis of numerous novel curcumin analogs, necessitating evaluation for their therapeutic potential. The selectivity of the top-scoring compounds was assessed through molecular docking studies and molecular dynamics simulations to determine their binding affinity to PLpro. As a result, we identified 20 potential, selective PLpro inhibitors, from which the top two compounds (THA111 and THHGV6) were selected based on their binding free energy values towards PLpro as estimated by MM-PBSA calculations. These selected candidates demonstrate promising activity against the protein, with binding free energy values ranging from approximately −105 to −108 kJ/mol, and largely adopt a similar binding mode to known noncovalent SARS-CoV-2 PLpro inhibitors (GRL0617 = −100.98 kJ/mol). We further propose these two most promising compounds for future in vitro evaluation. The findings for the top potential PLpro inhibitors have been deposited in a database (Curcumin Research Center) to aid research on anti-SARS-CoV-2 drugs.
Keywords
SARS-CoV-2 PLpro, Curcumin analogs, COVID-19 therapy, Virtual drug screening, Computational investigations
Introduction
Due to the rapid and widespread transmission rates, the World Health Organization (WHO) officially classified coronavirus disease 2019 (COVID-19) as a global health emergency and declared it a pandemic on March 11, 2020 (
SARS-CoV-2 belongs to the Coronaviridae family and is an enveloped positive-sense RNA virus (
The SARS-CoV-2 PLpro enzyme plays a crucial role in viral replication and in dampening the host immune response, making it an attractive target for intervention (
At present, numerous researchers have indicated the potential of plant-derived chemical compounds in combating SARS-CoV-2 infection, which could potentially prevent the onset or severity of COVID-19. Among these compounds, curcumin, the primary polyphenolic component found in turmeric, has garnered considerable attention due to its diverse biological effects, including its anti-tumor, anti-inflammatory, immunomodulatory, antioxidant, and antimicrobial properties (
Interestingly, previous in vitro studies have shown that post-infection treatment with curcumin at a concentration of 10 µg/mL exhibits significant antiviral effects against SARS-CoV-2, with inhibition rates of 99% and 99.8% against the DG614 strain and Delta variant respectively (
In this study, we aimed to discover new, potent, noncovalent, and specific inhibitors of PLpro. These compounds are anticipated to exhibit greater binding affinity to SARS-CoV-2 PLpro compared to existing inhibitors. To accomplish this objective, we conducted thorough in silico screening, a modern and efficient approach in drug design. We placed a strong emphasis on the accuracy of our predictions by extensively validating the techniques utilized to ensure their applicability to our project. Consequently, we employed a variety of computational methods, merging both ligand-based and structure-based strategies. Initially, our focus was on curcumin analogs, comprising a total of 20 compounds synthesized successfully in our laboratory. Subsequently, we assessed the binding affinities of selected molecules to SARS-CoV-2 PLpro through molecular docking and molecular dynamics simulations. These findings have been archived in a publicly accessible database to facilitate future research endeavors aimed at combating the COVID-19 pandemic.
Materials and methods
Preparation and optimization of curcumin analogs
The structural configurations of the compounds intended for the study were derived from both two-dimensional and three-dimensional representations generated using ChemDraw Professional 16.0 and Chem3D 16.0 software (
Preparation of SARS-CoV-2 papain-like protease (PLpro) macromolecules
The three-dimensional configuration of the SARS-CoV-2 papain-like protease (PLpro) receptor macromolecule was acquired from the Protein Data Bank (PDB) website via the URL https://www.rcsb.org/structure/3e9s (
Molecular docking studies
Molecular docking investigations of curcumin derivative compounds against SARS-CoV-2 PLpro macromolecules were conducted using AutoDock 4.2 to explore potential interactions between these curcumin analog compounds and SARS-CoV-2 PLpro macromolecules (
ADMET properties of curcumin analogs
The pharmacological and pharmacokinetic characteristics of all curcumin derivative compounds were assessed through the SwissADME (
PASS identification of curcumin analogs
The PASS website was employed for forecasting the pharmacological and biological traits of the substances (
Molecular dynamics simulations
Molecular dynamics simulations lasting 100 ns were conducted utilizing Gromacs 2016.3 with the AMBER99SB-ILDN force field, which enhances the accuracy of MD simulations by incorporating the Improved Lipophilic Efficiency Descriptors for Nucleic Acids (ILDN) parameter for nucleic acids (
Binding free energy MM-PBSA calculation
The Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) calculations were conducted using the g_mmpbsa package, which is integrated into the Gromacs 2016.3 software (
Results and discussion
Molecular docking studies
The initial stage involves conducting molecular docking simulations between curcumin analogs and SARS-CoV-2 papain-like protease (PLpro) macromolecules using AutoDock 4.2 equipped with the Lamarckian Genetic Algorithm (LGA). Molecular docking serves as a computational approach to elucidate the interactions between a specific chemical compound (ligand) and a protein (target) (
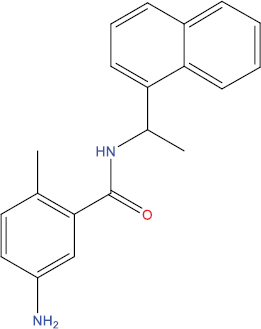
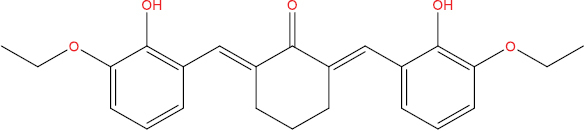
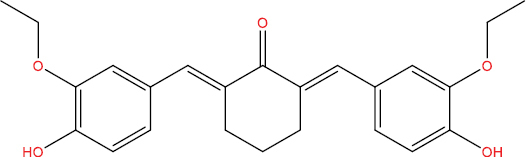
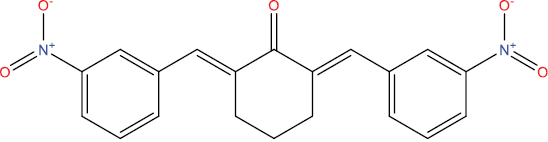
The outcomes of molecular docking investigations reveal that all curcumin analog compounds exhibit favorable affinity towards SARS-CoV-2 PLpro macromolecules. However, only five derivative compounds, namely A108, A113, A146, THA113, and THA146, demonstrate superior affinity compared to native ligands (GRL0617) (Table
The free energy of binding between curcumin analogs and SARS-CoV-2 PLpro macromolecules.
| Compound Molecule | Molecular Structure | Binding Free Energy |
|---|---|---|
| Native (GRL0617) |
|
−10.05 kcal/mol |
| A102 |
|
−8.01 kcal/mol |
| A103 |
|
−7.27 kcal/mol |
| A104 |
|
−8.57 kcal/mol |
| A108 |
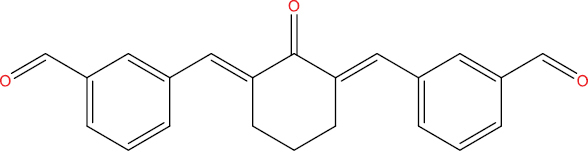
|
−9.22 kcal/mol |
| A111 |
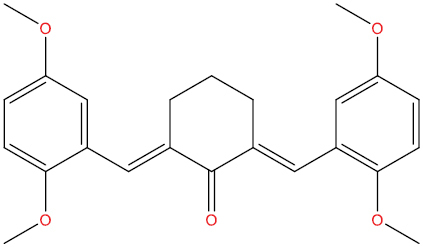
|
−7.90 kcal/mol |
| A113 |
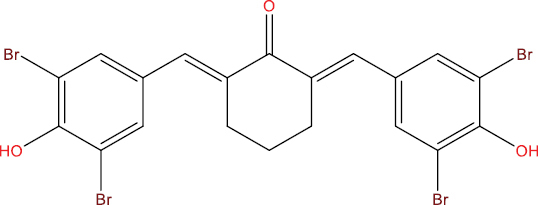
|
−9.85 kcal/mol |
| A129 |
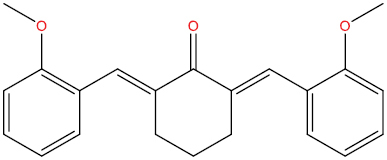
|
−8.52 kcal/mol |
| A146 |
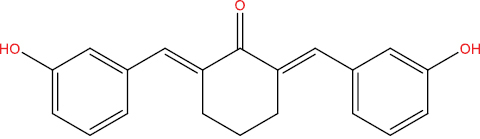
|
−9.96 kcal/mol |
| HGV5 |
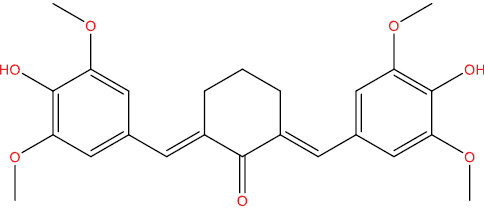
|
−8.18 kcal/mol |
| HGV6 |
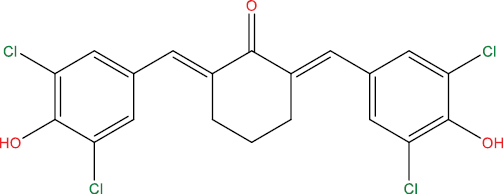
|
−8.65 kcal/mol |
| THA102 |
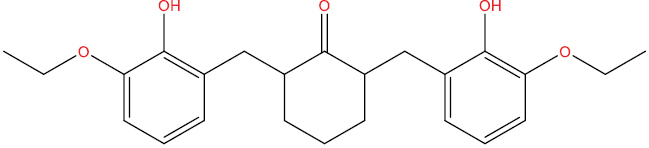
|
−7.83 kcal/mol |
| THA103 |
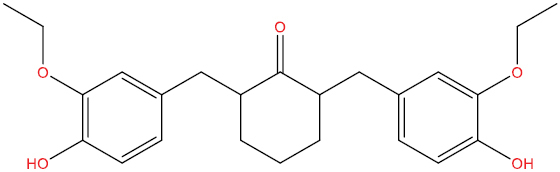
|
−7.61 kcal/mol |
| THA104 |
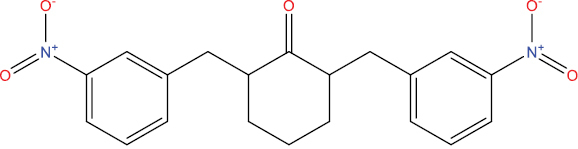
|
−8.83 kcal/mol |
| THA108 |
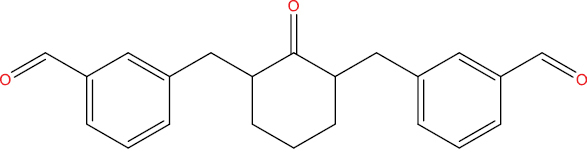
|
−8.89 kcal/mol |
| THA111 |
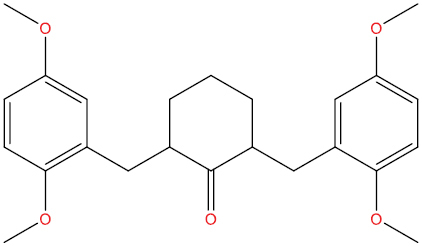
|
−7.15 kcal/mol |
| THA113 |
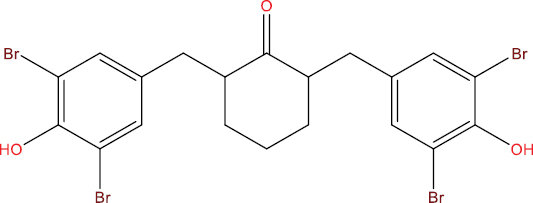
|
−9.29 kcal/mol |
| THA129 |
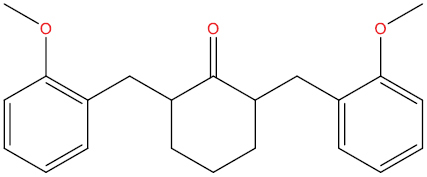
|
−8.14 kcal/mol |
| THA146 |
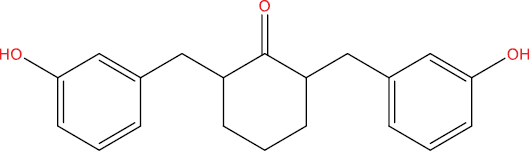
|
−9.56 kcal/mol |
| THHGV5 |
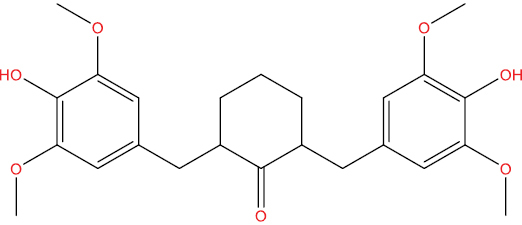
|
−7.47 kcal/mol |
| THHGV6 |
|
−8.93 kcal/mol |
In Fig.

It’s intriguing that having two symmetrically linked 1,3-dicarbonyl or α, β unsaturated carbonyl units facilitates binding with DNA, protein sites, and metals through a well-established process known as keto-enol tautomerism (

ADMET properties of curcumin analogs
Physiological and pharmacokinetic traits play a critical role in the selection and advancement of drug-like substances. Compounds that successfully undergo screening for physicochemical and ADMET properties stand a better chance of achieving clinical success. The pkCSM platform computes physicochemical and ADMET parameters for all compounds chosen through docking screening. For every curcumin derivative compound, a range of ADMET parameters is evaluated concurrently with PAINS screening. This process identifies twenty compounds possessing outstanding physicochemical characteristics and devoid of any PAINS patterns (
The ADMET criteria of the curcumin analogs utilizing the pkCSM Web tool.
| Compound Molecule | Molecular Structure | GI Absorption | Water Solubility (log mol/L) | BBB Permeability (log BB) | CYP2D6 Inhibitor | Renal OCT2 Substrate | AMES Toxicity |
|---|---|---|---|---|---|---|---|
| A102 |
|
High | −3.91 | Yes | Yes | No | No |
| A103 |
|
High | −3.91 | Yes | Yes | No | No |
| A104 |
|
High | −4.75 | No | No | No | No |
| A108 |
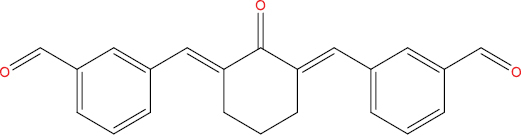
|
High | −4.31 | Yes | Yes | No | No |
| A111 |
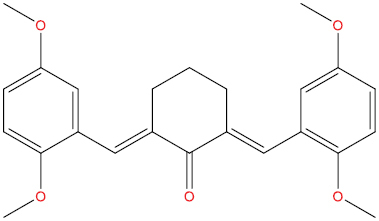
|
High | −4.13 | Yes | Yes | No | No |
| A113 |
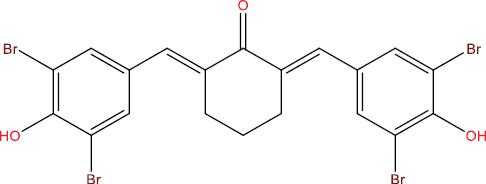
|
High | −5.76 | No | No | No | No |
| A129 |
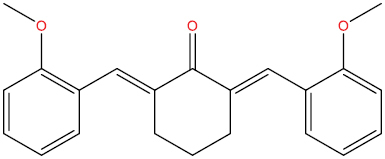
|
High | −4.67 | Yes | Yes | No | No |
| A146 |
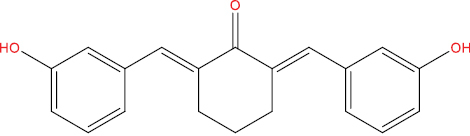
|
High | −3.95 | Yes | No | No | No |
| HGV5 |
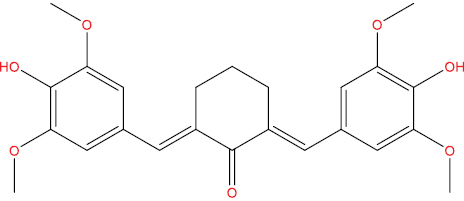
|
High | −3.10 | No | No | No | No |
| HGV6 |
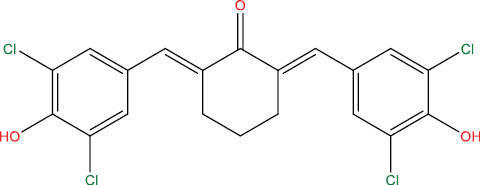
|
High | −4.89 | Yes | No | No | No |
| THA102 |
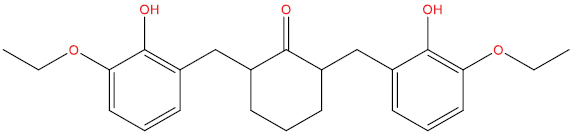
|
High | −3.91 | Yes | Yes | No | No |
| THA103 |
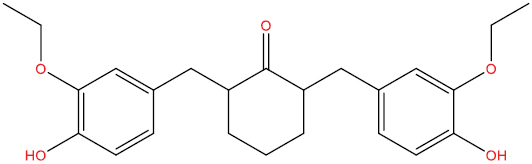
|
High | −3.91 | Yes | Yes | No | No |
| THA104 |
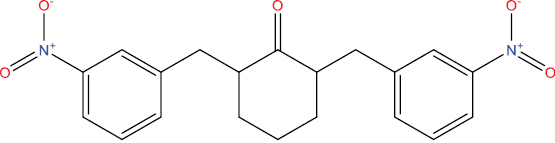
|
High | −4.75 | No | No | No | No |
| THA108 |
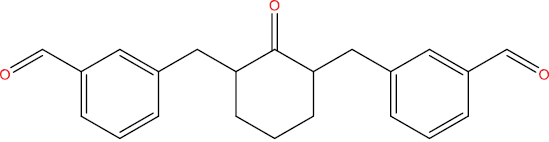
|
High | −4.31 | Yes | Yes | No | No |
| THA111 |
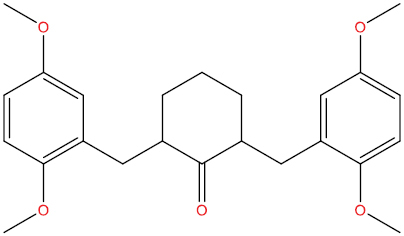
|
High | −4.13 | Yes | Yes | No | No |
| THA113 |
|
High | −5.76 | Yes | No | No | No |
| THA129 |
|
High | −4.76 | Yes | Yes | No | No |
| THA146 |
|
High | −3.95 | Yes | No | No | No |
| THHGV5 |
|
High | −3.10 | No | No | No | No |
| THHGV6 |
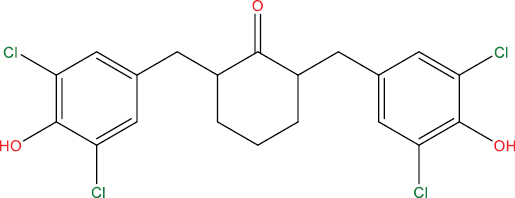
|
High | −4.89 | Yes | No | No | No |
PASS identification of curcumin analogs
The PASS server holds an extensive dataset for training, encompassing diverse bioactive compounds and their associations between structure and activity derived from a range of clinical and preclinical investigations. Using this dataset, the PASS server predicts the biological activity of chemical compounds (
The primary 20 pertinent biological characteristics of the clarified curcumin analog compounds.
| Compound Molecule | Molecular Structure | Possible Activity (Pa) | Possible Inactivity (Pi) | Biological Activity |
|---|---|---|---|---|
| A102 |
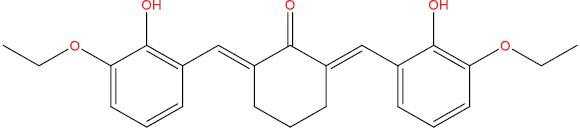
|
0.416 | 0,074 | Antiviral (Rhinovirus) |
| 0.337 | 0.062 | Antiviral (Adenovirus) | ||
| 0.364 | 0.143 | Antiviral (Picornavirus) | ||
| 0.286 | 0.138 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.212 | 0.147 | 3C-like protease (Human coronavirus) inhibitor | ||
| A103 |
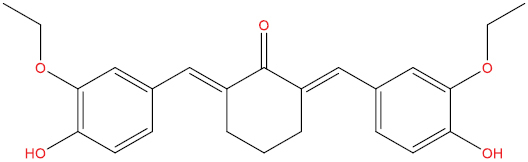
|
0.425 | 0.067 | Antiviral (Rhinovirus) |
| 0.371 | 0.042 | Antiviral (Adenovirus) | ||
| 0.330 | 0.183 | Antiviral (Picornavirus) | ||
| 0.238 | 0.096 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.277 | 0.148 | Simian immunodeficiency virus proteinase inhibitor | ||
| A104 |
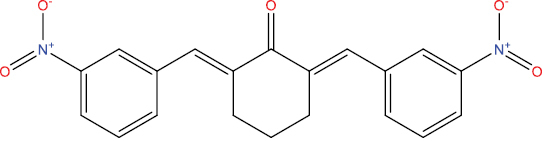
|
0.552 | 0.031 | Antiviral (Picornavirus) |
| 0.488 | 0.023 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.459 | 0.012 | Antiviral (Adenovirus) | ||
| 0.396 | 0.004 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.195 | 0.166 | Antiviral (Poxvirus) | ||
| A108 |
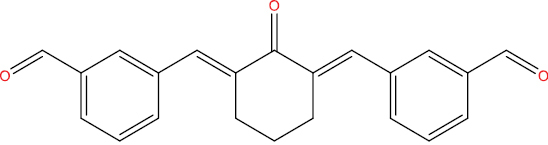
|
0.285 | 0.035 | 3C-like protease (Human coronavirus) inhibitor |
| 0.315 | 0.077 | Antiviral (Adenovirus) | ||
| 0.326 | 0.099 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.312 | 0.207 | Antiviral (Picornavirus) | ||
| A111 |
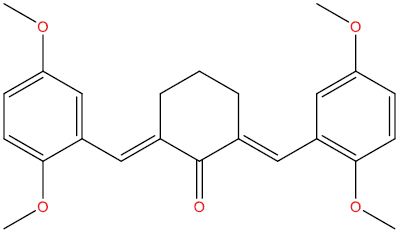
|
0.328 | 0.097 | Simian immunodeficiency virus proteinase inhibitor |
| 0.272 | 0.047 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.285 | 0.102 | Antiviral (Adenovirus) | ||
| 0.342 | 0.167 | Antiviral (Picornavirus) | ||
| A113 |
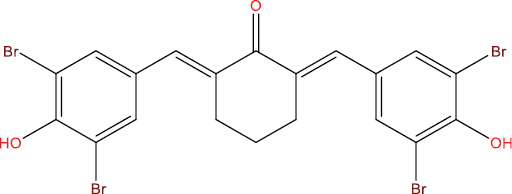
|
0.546 | 0.004 | Antiviral (Adenovirus) |
| 0.284 | 0.037 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.285 | 0.138 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.210 | 0.144 | Antiviral (Poxvirus) | ||
| 0.279 | 0.263 | Antiviral (Picornavirus) | ||
| A129 |
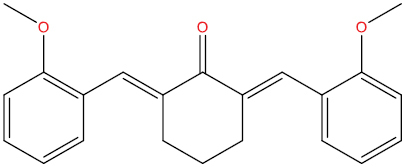
|
0.376 | 0.130 | Antiviral (Picornavirus) |
| 0.376 | 0.130 | Antiviral (Picornavirus) | ||
| 0.301 | 0.088 | Antiviral (Adenovirus) | ||
| 0.316 | 0.107 | Simian immunodeficiency virus proteinase inhibitor | ||
| A146 |
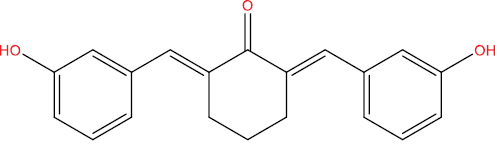
|
0.403 | 0.028 | Antiviral (Adenovirus) |
| 0.320 | 0.015 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.349 | 0.081 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.381 | 0.126 | Antiviral (Picornavirus) | ||
| 0.194 | 0.168 | Antiviral (Poxvirus) | ||
| HGV5 |
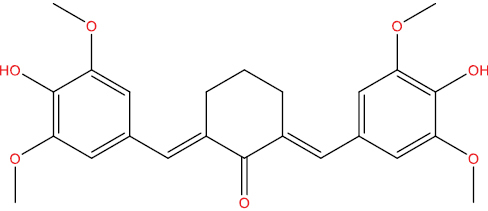
|
0.371 | 0.043 | Antiviral (Adenovirus) |
| 0.289 | 0.032 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.322 | 0.193 | Antiviral (Picornavirus) | ||
| 0.266 | 0.162 | Simian immunodeficiency virus proteinase inhibitor | ||
| HGV6 |
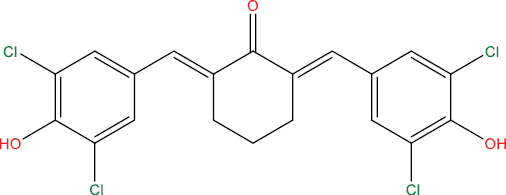
|
0.417 | 0.097 | Antiviral (Picornavirus) |
| 0.344 | 0.057 | Antiviral (Adenovirus) | ||
| 0.289 | 0.032 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.285 | 0.138 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.187 | 0.180 | Antiviral (Poxvirus) | ||
| THA102 |
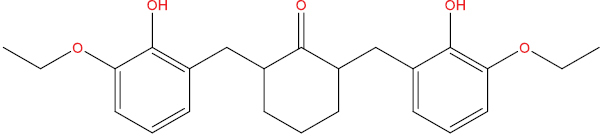
|
0.523 | 0.041 | Antiviral (Picornavirus) |
| 0.431 | 0.062 | Antiviral (Rhinovirus) | ||
| 0.274 | 0.113 | Antiviral (Adenovirus) | ||
| 0.276 | 0.150 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.229 | 0.111 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.214 | 0.138 | Antiviral (Poxvirus) | ||
| THA103 |
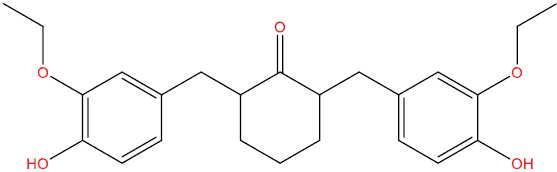
|
0.531 | 0.038 | Antiviral (Picornavirus) |
| 0.439 | 0.056 | Antiviral (Rhinovirus) | ||
| 0.247 | 0.081 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.261 | 0.127 | Antiviral (Adenovirus) | ||
| 0.267 | 0.161 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.201 | 0.157 | Antiviral (Poxvirus) | ||
| THA104 |
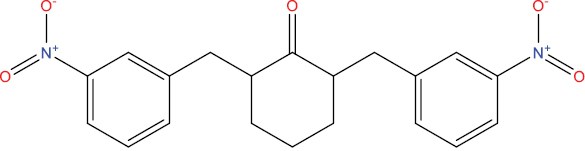
|
0.713 | 0.005 | Antiviral (Picornavirus) |
| 0.476 | 0.026 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.410 | 0.004 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.359 | 0.049 | Antiviral (Adenovirus) | ||
| 0.227 | 0.120 | Antiviral (Poxvirus) | ||
| 0.026 | 0.007 | Protease (Human cytomegalovirus) inhibitor | ||
| THA108 |
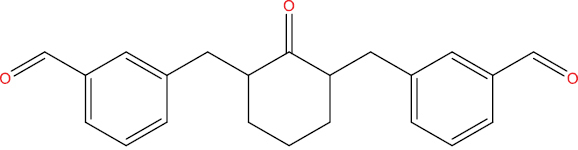
|
0.510 | 0.046 | Antiviral (Picornavirus) |
| 0.294 | 0.028 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.315 | 0.108 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.207 | 0.198 | Antiviral (Adenovirus) | ||
| THA111 |
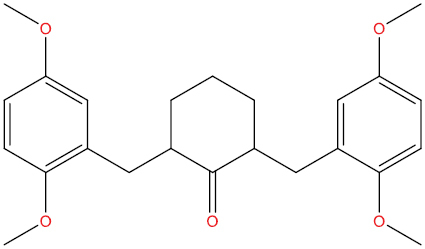
|
0.499 | 0.051 | Antiviral (Picornavirus) |
| 0.289 | 0.032 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.317 | 0.106 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.315 | 0.222 | Antiviral (Rhinovirus) | ||
| 0.225 | 0.171 | Antiviral (Adenovirus) | ||
| 0.188 | 0.179 | Antiviral (Poxvirus) | ||
| THA113 |
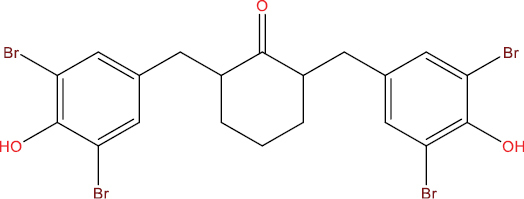
|
0.453 | 0.013 | Antiviral (Adenovirus) |
| 0.467 | 0.066 | Antiviral (Picornavirus) | ||
| 0.293 | 0.029 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.242 | 0.103 | Antiviral (Poxvirus) | ||
| 0.275 | 0.150 | Simian immunodeficiency virus proteinase inhibitor | ||
| THA129 |
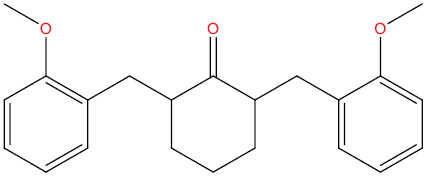
|
0.536 | 0.037 | Antiviral (Picornavirus) |
| 0.298 | 0.026 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.306 | 0.116 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.239 | 0.152 | Antiviral (Adenovirus) | ||
| 0.203 | 0.154 | Antiviral (Poxvirus) | ||
| 0.285 | 0.285 | Antiviral (Rhinovirus) | ||
| THA146 |
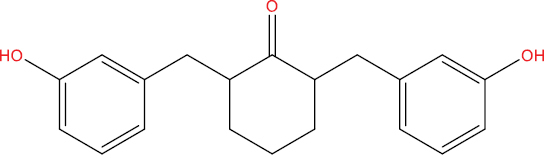
|
0.585 | 0.023 | Antiviral (Picornavirus) |
| 0.330 | 0.012 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.338 | 0.088 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.295 | 0.093 | Antiviral (Adenovirus) | ||
| 0.226 | 0.122 | Antiviral (Poxvirus) | ||
| THHGV5 |
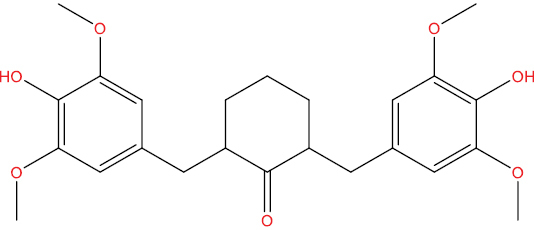
|
0.523 | 0.041 | Antiviral (Picornavirus) |
| 0.298 | 0.026 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.260 | 0.128 | Antiviral (Adenovirus) | ||
| 0.257 | 0.175 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.203 | 0.154 | Antiviral (Poxvirus) | ||
| 0.292 | 0.270 | Antiviral (Rhinovirus) | ||
| THHGV6 |
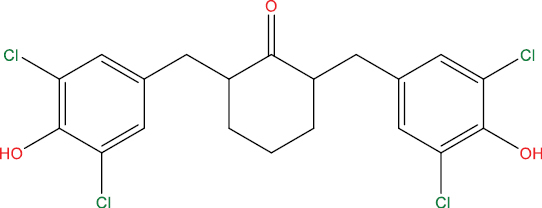
|
0.616 | 0.016 | Antiviral (Picornavirus) |
| 0.298 | 0.026 | 3C-like protease (Human coronavirus) inhibitor | ||
| 0.275 | 0.150 | Simian immunodeficiency virus proteinase inhibitor | ||
| 0.219 | 0.132 | Antiviral (Poxvirus) | ||
| 0.233 | 0.160 | Antiviral (Adenovirus) |
Binding free energy MM-PBSA calculation
The subsequent step involves conducting molecular dynamics simulations between all curcumin-analog compounds and the SARS-CoV-2 PLpro macromolecules. Molecular dynamics simulations represent a computational technique employed to assess molecular behavior within biological systems (
The Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method is employed to assess the interaction energy between molecules and their surroundings within molecular dynamics simulations. This approach integrates principles from molecular mechanics and Poisson-Boltzmann theory to compute the interaction energy of molecules within their environment. MM-PBSA evaluates the interaction energy between molecules by employing molecular mechanics calculations to determine the potential energy within a molecule, which represents the energy required for forming its molecular structure (
Based on the results obtained from the MM-PBSA method for binding-free energy calculations, only five curcumin analog compounds demonstrated superior affinity and stability compared to the native ligands (GRL0617) during the 100 ns simulation in molecular dynamics interactions. Among these, A102, THA102, THA104, THA111, and THHGV6 exhibited the highest affinity stability, with binding free energy values of −108.975 kJ/mol, −108.931 kJ/mol, −108.975 kJ/mol, −106.188 kJ/mol, and −105.023 kJ/mol, respectively (Table
The binding free energy MM-PBSA calculation of curcumin analogs with SARS-CoV-2 PLpro macromolecules.
| Compound Molecule | ∆Evdw (kJ/mol) | ∆Eele (kJ/mol) | ∆GPB (kJ/mol) | ∆GNP (kJ/mol) | ∆GBind (kJ/mol) |
|---|---|---|---|---|---|
| Native (GRL0617) | −149.05 | −43.41 | 107.66 | −16.17 | −100.98 |
| A102 | −163.03 | −32.83 | 103.36 | −16.47 | −108.98 |
| A103 | −104.99 | −13.20 | 60.00 | −12.46 | −70.65 |
| A104 | −124.74 | −21.12 | 100.84 | −12.90 | −57.91 |
| A108 | −115.35 | −13.82 | 96.43 | −12.88 | −45.61 |
| A111 | −206.94 | −30.14 | 171.81 | −20.06 | −85.33 |
| A113 | −146.69 | −69.31 | 156.90 | −13.61 | −72.70 |
| A129 | −126.14 | −4.07 | 75.85 | −13.87 | −68.24 |
| A146 | −81.52 | −17.58 | 77.34 | −9.68 | −31.44 |
| HGV5 | −155.07 | −40.02 | 125.77 | −17.70 | −87.03 |
| HGV6 | −137.38 | −32.72 | 119.76 | −16.32 | −66.65 |
| THA102 | −180.07 | −24.07 | 113.11 | −17.89 | −108.93 |
| THA103 | −107.48 | −22.78 | 77.19 | −12.62 | −65.69 |
| THA104 | −163.03 | −32.83 | 103.36 | −16.47 | −108.98 |
| THA108 | −116.88 | −14.94 | 79.21 | −13.56 | −66.17 |
| THA111 | −170.04 | −28.31 | 110.86 | −18.70 | −106.19 |
| THA113 | −107.50 | −12.34 | 58.94 | −11.15 | −72.06 |
| THA129 | −112.04 | −15.79 | 66.62 | −12.70 | −73.91 |
| THA146 | −129.58 | −31.90 | 100.00 | −14.88 | −76.37 |
| THHGV5 | −151.65 | −36.58 | 104.74 | −15.67 | −99.16 |
| THHGV6 | −155.60 | −29.61 | 97.08 | −16.89 | −105.02 |
Interaction stability in molecular dynamics simulations
The purpose of trajectory visualization in molecular dynamics simulation is to enhance comprehension of particle behavior within the molecular system under investigation. In molecular dynamics simulations, particles, whether atoms or molecules, are discrete entities influenced by forces from other particles within the system. By visualizing particle trajectories, we can observe how particles interact and how alterations in simulation conditions, such as temperature or density, impact particle behavior. These visualizations aid in recognizing patterns and trends in particle motion, illustrating the correlation between particle movement and evolving system conditions, and portraying outcomes across various simulation scenarios.
Several factors can lead to the displacement of a compound from its binding site during molecular dynamics simulations. Primarily, the instability of the interaction potential energy between curcumin analog compounds and the binding sites of SARS-CoV-2 PLpro macromolecules plays a significant role. This potential energy, which governs the stability of the compound at the binding site, is determined by the molecular interaction potential employed in the simulation. Certain potential interactions promote the compound’s stability at the binding site, while others may facilitate its movement away from it. Fig.

Furthermore, molecular dynamics simulations also consider the thermal transfer of atoms participating in interactions. If atoms involved in the interaction are sufficiently large during the heat transfer simulation, they may induce the compound to shift away from the binding site. To delve deeper into the molecular interactions during the 100 ns molecular dynamics simulations, Root Mean Square Deviation (RMSD) and Root Mean Square Fluctuation (RMSF) analyses were conducted. RMSD quantifies the average disparity between the protein’s true conformation (derived from the crystal structure) and the conformation acquired from molecular dynamics simulations. It is computed by measuring the mean distance between atoms in the two conformations. A low RMSD value suggests a close resemblance between the conformation from molecular dynamics simulations and the actual conformation. Conversely, RMSF assesses the average fluctuation of atomic positions in dynamic conformations obtained from molecular dynamics simulations. RMSF is determined by computing the average distance of each atom from its mean position in the dynamic conformation. A high RMSF value indicates substantial atomic movement in the dynamic conformation obtained from molecular dynamics simulations (
The RMSD chart displayed in Fig.

Distribution of molecule movement during molecular dynamics simulations
Graphical examination of the radius of gyration (Rg) and solvent accessible surface area (SASA) is essential to validate the fluctuations observed in the results obtained from molecular dynamics simulations for each curcumin analog compound. Rg serves as a parameter to gauge the size and distribution of mass within molecules or molecular assemblies (
Fig.

Atomic distribution in molecular dynamics simulations systems
The radial distribution function (RDF) serves as a metric utilized to assess the dispersion pattern of atoms or molecules within a system. In molecular dynamics simulations, RDF is employed to gauge the spatial arrangement of atoms within the simulated protein. This calculation involves dividing the mean distance between two distinct types of atoms by the average distance of identical atoms (
Compound A102 exhibits a consistently stable and adaptable atomic dispersion throughout the simulations (Fig.

Strength and geometry of hydrogen bonds in molecular dynamics simulations
Moreover, an assessment of the stability of the ligand-protein hydrogen bonds established during the molecular dynamics simulations was conducted. This entails scrutinizing the hydrogen bond dynamics derived from the simulations, encompassing the quantification of their quantity, strength, and configuration. This analysis involves parsing the atomic coordinate datasets generated from the simulations and aligning them with predefined geometric criteria for hydrogen bonding. The tally of hydrogen bonds formed serves as a metric to gauge system stability, with fluctuations indicating alterations in molecular conformation. Additionally, the vigor of the hydrogen bond is appraised through an examination of its potential energy, while its geometry is assessed by scrutinizing the distance and angle between the hydrogen and bonded atoms (
Based on the hydrogen bond occupancy data presented in Table
Percentage of hydrogen bonds formed by curcumin analogs with SARS-CoV-2 PLpro macromolecules.
| Compound Molecule | Donor | Acceptor | Occupancy | Total Occupancy |
|---|---|---|---|---|
| A102 | TYR266 | A102 | 0.60% | 11.28% |
| LYS159 | A102 | 0.10% | ||
| GLN271 | A102 | 0.30% | ||
| GLN271 | A102 | 0.90% | ||
| GLN271 | A102 | 9.18% | ||
| A102 | ASP166 | 0.20% | ||
| THA102 | THA102 | ASP166 | 0.10% | 6.29% |
| GLN271 | THA102 | 0.50% | ||
| TYR270 | THA102 | 5.39% | ||
| GLN271 | THA102 | 0.30% | ||
| THA104 | GLN271 | THA104 | 14.37% | 14.47% |
| GLN271 | THA104 | 0.10% | ||
| THA111 | GLN271 | THA111 | 10.38% | 21.76% |
| GLN271 | THA111 | 1.50% | ||
| GLN271 | THA111 | 5.69% | ||
| TYR266 | THA111 | 4.19% | ||
| THHGV6 | THHGV6 | GLU169 | 1.20% | 23.36% |
| GLN271 | THHGV6 | 22.16% |
Fig.

Conclusions
The simulations indicate that overall, curcumin analog compounds exhibit favorable binding to SARS-CoV-2 papain-like protease (PLpro) macromolecules. However, THA111 and THHGV6 compounds demonstrated superior stability and affinity for SARS-CoV-2 PLpro macromolecules in both molecular dynamics simulations and MM-PBSA binding-free energy calculations. Consequently, these two compounds emerge as promising therapeutic candidates for combating COVID-19.
Acknowledgments
Author thanks the Curcumin Research Centre, Faculty of Pharmacy, Universitas Gadjah Mada, for providing the database of curcumin analog compounds used in this study.
This research received no external funding.
References
- Abduljalil JM, Abduljalil BM (2020) Epidemiology, genome, and clinical features of the pandemic SARS-CoV-2: a recent view. New Microbes and New Infections 35: 100672. https://doi.org/10.1016/j.nmni.2020.100672
- Abraham M, Hess B, van der Spoel D, Lindahl E (2015) The GROMACS development team, GROMACS user manual. Version 5.0.7.
- Adamczak A, Ożarowski M, Karpiński TM (2020) Curcumin, a natural antimicrobial agent with strain-specific activity. Pharmaceuticals 13(7): 153. https://doi.org/10.3390/ph13070153
- Angamuthu D, Purushothaman I, Kothandan S, Swaminathan R (2019) Antiviral study on Punica granatum L., Momordica charantia L., Andrographis paniculata Nees, and Melia azedarach L., to Human Herpes Virus-3. European Journal of Integrative Medicine 28(4): 98–108. https://doi.org/10.1016/j.eujim.2019.04.008
- Aragones JL, Noya EG, Valeriani C, Vega C (2013) Free energy calculations for molecular solids using GROMACS. Journal of Chemical Physics 139: 034104. https://doi.org/10.1063/1.4812362
- Ariyanto EF, Shalannandia WA, Lantika UA, Fakih TM, Ramadhan DSF, Gumilar AN, Permana FK, Rahmah AN, Atik N, Khairani AF (2023) Anthocyanin-containing purple sweet potato (Ipomoea batatas L.) Synbiotic Yogurt Inhibited 3T3-L1 Adipogenesis by Suppressing White Adipocyte-Specific Genes. Journal of Experimental Pharmacology 15: 217–230. https://doi.org/10.2147/JEP.S405433
- Aulifa DL, Al Shofwan AA, Megantara S, Fakih TM, Budiman A (2024) Elucidation of Molecular Interactions Between Drug–Polymer in Amorphous Solid Dispersion by a Computational Approach Using Molecular Dynamics Simulations. Advances and Applications in Bioinformatics and Chemistry 17: 1–19. https://doi.org/10.2147/AABC.S441628
- Bianchi E, Pessi A (2002) Inhibiting viral proteases: Challenges and opportunities. Biopolymers 66(2): 101–114. https://doi.org/10.1002/bip.10230
- BIOVIA (2017) Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017. Dassault Systèmes San Diego.
- Brown T (2014) ChemDraw. The Science Teacher 81. https://doi.org/10.12968/prtu.2014.1.37.65
- Cheung CKM, Law MF, Lui GCY, Wong SH (2021) Coronavirus Disease 2019 (COVID-19): A Haematologist’s Perspective. Acta Haematologica 144(1): 10–23. https://doi.org/10.1159/000510178
- Daina A, Michielin O, Zoete V (2017) SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 7: 42717. https://doi.org/10.1038/srep42717
- Das S, Sarmah S, Lyndem S, Singha Roy A (2021) An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics 39(9): 3347–3357. https://doi.org/10.1080/07391102.2020.1763201
- Deshmukh TR, Krishna VS, Sriram D, Sangshetti JN, Shingate BB (2020) Synthesis and bioevaluation of α,α’-bis(1H-1,2,3-triazol-5-ylmethylene) ketones. Chemical Papers 74(3): 809–820. https://doi.org/10.1007/s11696-019-00908-5
- Fakih TM (2023) Molecularly imprinted polymer-based sensors for identification volatile compounds in pharmaceutical products: in silico rational design. Journal of Biomolecular Structure and Dynamics 1–11. https://doi.org/10.1080/07391102.2023.2252090
- Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee MH, Wood J, O’Dea MR, Dutton S, Shamardani K, Nwangwu K, Mancusi R, Yalçın B, Taylor KR, Acosta-Alvarez L, Malacon K, Keough MB, Ni L, Woo PJ, Contreras-Esquivel D, Toland AMS, Gehlhausen JR, Klein J, Takahashi T, Silva J, Israelow B, Lucas C, Mao T, Peña-Hernández MA, Tabachnikova A, Homer RJ, Tabacof L, Tosto-Mancuso J, Breyman E, Kontorovich A, McCarthy D, Quezado M, Vogel H, Hefti MM, Perl DP, Liddelow S, Folkerth R, Putrino D, Nath A, Iwasaki A, Monje M (2022) Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell 185(14): 2452–2468. https://doi.org/10.1016/j.cell.2022.06.008
- Forli W, Halliday S, Belew R, Olson A (2012) AutoDock Version 4.2. Citeseer.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JAJ, Peralta PE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Ortiz J V, Cioslowski J, Fox DJ (2009) Gaussian 09, revision C.01; Gaussian Inc.: Wallingford, CT.
- Gil RM, Marcelin JR, Zuniga-Blanco B, Marquez C, Mathew T, Piggott DA (2020) COVID-19 pandemic: Disparate health impact on the hispanic/latinx population in the United States. Journal of Infectious Diseases 222(10): 1592–1595. https://doi.org/10.1093/infdis/jiaa474
- Hidayat AF, Fakih TM (2021) Self-assembly of black cumin oil-based nanoemulsion on various surfactants: A molecular dynamics study. Makara Journal of Science 25(4): 258–264. https://doi.org/10.7454/mss.v25i4.1267
- Hikmawati D, Fakih TM, Sutedja E, Dwiyana RF, atik N, Ramadhan DSF (2022) Pharmacophore-guided virtual screening and dynamic simulation of Kallikrein-5 inhibitor: Discovery of potential molecules for rosacea therapy. Informatics in Medicine Unlocked 28: 100844. https://doi.org/10.1016/j.imu.2022.100844
- Hulce KR, Jaishankar P, Lee GM, Bohn MF, Connelly EJ, Wucherer K, Ongpipattanakul C, Volk RF, Chuo SW, Arkin MR, Renslo AR, Craik CS (2022) Inhibiting a dynamic viral protease by targeting a non-catalytic cysteine. Cell Chemical Biology 29(5): 785–798. https://doi.org/10.1016/j.chembiol.2022.03.007
- Islam S, Hosen MA, Ahmad S, ul Qamar MT, Dey S, Hasan I, Fujii Y, Ozeki Y, Kawsar SMA (2022) Synthesis, antimicrobial, anticancer activities, PASS prediction, molecular docking, molecular dynamics and pharmacokinetic studies of designed methyl α-D-glucopyranoside esters. Journal of Molecular Structure 1260: 132761. https://doi.org/10.1016/j.molstruc.2022.132761
- Kaur G, Kaur M, Bansal M (2021) New insights of structural activity relationship of curcumin and correlating their efficacy in anticancer studies with some other similar molecules. American Journal of Cancer Research 11(8): 3755–3765.
- Kong A, Oh J-E, Lam T (2021) Face mask effects during COVID-19: perspectives of managers, practitioners and customers in the hotel industry. International Hospitality Review 35(2): 195–207. https://doi.org/10.1108/IHR-07-2020-0025
- Kotha RR, Luthria DL (2019) Curcumin: Biological, pharmaceutical, nutraceutical, and analytical aspects. Molecules 24(16): 2930. https://doi.org/10.3390/molecules24162930
- Kotni Meena NC (2015) QM/MM Docking Strategy and Prime/MM-GBSA Calculation of Celecoxib Analogues as N-myristoyltransferase Inhibitors. Virology & Mycology 4(1): 1–8. https://doi.org/10.4172/2161-0517.1000141
- Kumar N, Kaur K, Bedi PMS (2023) Hybridization of molecular docking studies with machine learning based QSAR model for prediction of xanthine oxidase activity. Computational and Theoretical Chemistry 1227: 114262. https://doi.org/10.1016/j.comptc.2023.114262
- Li D, Luan J, Zhang L (2021) Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochemical and Biophysical Research Communications 538: 72–79. https://doi.org/10.1016/j.bbrc.2020.11.083
- Linda Laksmiani NP, Febryana Larasanty LP, Jaya Santika AAG, Andika Prayoga PA, Kharisma Dewi AAI, Kristiara Dewi NPA (2020) Active compounds activity from the medicinal plants against SARS-CoV-2 using in silico assay. Biomedical and Pharmacology Journal 13(2): 873–881. https://doi.org/10.13005/bpj/1953
- Lohidashan K, Rajan M, Ganesh A, Paul M, Jerin J (2018) Pass and Swiss ADME collaborated in silico docking approach to the synthesis of certain pyrazoline spacer compounds for dihydrofolate reductase inhibition and antimalarial activity. Bangladesh Journal of Pharmacology 13: 23–29. https://doi.org/10.3329/bjp.v13i1.33625
- Marín-Palma D, Tabares-Guevara JH, Zapata-Cardona MI, Flórez-álvarez L, Yepes LM, Rugeles MT, Zapata-Builes W, Hernandez JC, Taborda NA (2021) Curcumin inhibits in vitro sars-cov-2 infection in vero e6 cells through multiple antiviral mechanisms. Molecules 26(22): 6900. https://doi.org/10.3390/molecules26226900
- Mishra A, Mulpuru V, Mishra N (2022) Exploring the mechanism of action of podophyllotoxin derivatives through molecular docking, molecular dynamics simulation and MM/PBSA studies. Journal of Biomolecular Structure and Dynamics 41(18): 8856–8865. https://doi.org/10.1080/07391102.2022.2138549
- Misran MRI Bin, Nunotani N, Tamura S, Imanaka N (2020) Enhancement of bromide ion conductivity in lanthanum oxybromide based solids by doping divalent zinc ion with high electronegativity. Journal of Asian Ceramic Societies 8(3): 925–929. https://doi.org/10.1080/21870764.2020.1793877
- Mohamed Thamby BF, Santhi VM, Ramalingam A (2023) Quantum chemical and experimental studies on the extraction of acid blue 80 and acid red 1 from their aquatic environment using tetrabutylammonium bromide based deep eutectic solvents. Journal of Dispersion Science and Technology 44(9): 1778–1787. https://doi.org/10.1080/01932691.2023.2195931
- Muchtaridi M, Triwahyuningtyas D, Muhammad Fakih T, Megantara S, Choi SB (2023) Mechanistic insight of α-mangostin encapsulation in 2-hydroxypropyl-β-cyclodextrin for solubility enhancement. Journal of Biomolecular Structure and Dynamics 42(6): 3223–3232. https://doi.org/10.1080/07391102.2023.2214237
- Nocito MC, De Luca A, Prestia F, Avena P, La Padula D, Zavaglia L, Sirianni R, Casaburi I, Puoci F, Chimento A, Pezzi V (2021) Antitumoral activities of curcumin and recent advances to improve its oral bioavailability. Biomedicines 9(10): 1476. https://doi.org/10.3390/biomedicines9101476
- Nurisyah , Ramadhan DSF, Dewi R, asikin A, Daswi DR, Adam A, Chaerunnimah , Sunarto , Rafika , Artati , Fakih TM (2024) Targeting EGFR allosteric site with marine-natural products of Clathria sp.: A computational approach. Current Research in Structural Biology 7: 100125. https://doi.org/10.1016/j.crstbi.2024.100125
- Osipiuk J, Azizi SA, Dvorkin S, Endres M, Jedrzejczak R, Jones KA, Kang S, Kathayat RS, Kim Y, Lisnyak VG, Maki SL, Nicolaescu V, Taylor CA, Tesar C, Zhang YA, Zhou Z, Randall G, Michalska K, Snyder SA, Dickinson BC, Joachimiak A (2021) Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nature Communications 12(1): 743. https://doi.org/10.1038/s41467-021-21060-3
- Pires DEV, Blundell TL, Ascher DB (2015) pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. Journal of Medicinal Chemistry 58(9): 4066–4072. https://doi.org/10.1021/acs.jmedchem.5b00104
- Pitaloka DAE, Ramadhan DSF, Arfan , Chaidir L, Fakih TM (2021) Docking-based virtual screening and molecular dynamics simulations of quercetin analogs as enoyl-acyl carrier protein reductase (Inha) inhibitors of mycobacterium tuberculosis. Scientia Pharmaceutica 89(2): 20. https://doi.org/10.3390/scipharm89020020
- Ramadhan DSF, Fakih TM, Arfan A (2020) Activity Prediction of Bioactive Compounds Contained in Etlingera elatior Against the SARS-CoV-2 Main Protease: An In Silico Approach. Borneo Journal of Pharmacy 3(4): 235–242. https://doi.org/10.33084/bjop.v3i4.1634
- Ramadhan DSF, Siharis F, Abdurrahman S, Isrul M, Fakih TM (2022) In silico analysis of marine natural product from sponge (Clathria Sp.) for their activity as inhibitor of SARS-CoV-2 Main Protease. Journal of Biomolecular Structure and Dynamics 40(22): 11526–11532. https://doi.org/10.1080/07391102.2021.1959405
- Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Baliji S, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD (2008) A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proceedings of the National Academy of Sciences of the United States of America 105(42): 16119–16124. https://doi.org/10.1073/pnas.0805240105
- Sencanski M, Perovic V, Milicevic J, Todorovic T, Prodanovic R, Veljkovic V, Paessler S, Glisic S (2022) Identification of SARS-CoV-2 Papain-like Protease (PLpro) Inhibitors Using Combined Computational Approach. ChemistryOpen 11(2): e202100248. https://doi.org/10.1002/open.202100248
- Shah V, Bhaliya J, Patel GM (2022) In silico docking and ADME study of deketene curcumin derivatives (DKC) as an aromatase inhibitor or antagonist to the estrogen-alpha positive receptor (Erα+): potent application of breast cancer. Structural Chemistry 33(2): 571–600. https://doi.org/10.1007/s11224-021-01871-2
- Smith MD, Rao JS, Segelken E, Cruz L (2015) Force-Field Induced Bias in the Structure of Aβ21-30: A Comparison of OPLS, AMBER, CHARMM, and GROMOS Force Fields. Journal of Chemical Information and Modeling 55(12): 2587–2595. https://doi.org/10.1021/acs.jcim.5b00308
- Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, Barnes S, Grizzle W, Miller D, Zhang HG (2010) A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Molecular Therapy 18(9): 1606–1614. https://doi.org/10.1038/mt.2010.105
- Tan H, Hu Y, Jadhav P, Tan B, Wang J (2022) Progress and Challenges in Targeting the SARS-CoV-2 Papain-like Protease. Journal of Medicinal Chemistry 65(11): 7561–7580. https://doi.org/10.1021/acs.jmedchem.2c00303
- Tomeh MA, Hadianamrei R, Zhao X (2019) A review of curcumin and its derivatives as anticancer agents. International Journal of Molecular Sciences 20(5): 1033. https://doi.org/10.3390/ijms20051033
- Urošević M, Nikolić L, Gajić I, Nikolić V, Dinić A, Miljković V (2022) Curcumin: Biological Activities and Modern Pharmaceutical Forms. Antibiotics 11(2): 135. https://doi.org/10.3390/antibiotics11020135
- Vijayakumar SD, Zakaria J, Ridzuan N (2022) Molecular dynamics approach on intermolecular interaction between n-icosane and gemini surfactant assisted nanoparticles. Petroleum Research 7(3): 366–371. https://doi.org/10.1016/j.ptlrs.2021.12.001
- van Vliet VJE, Huynh N, Palà J, Patel A, Singer A, Slater C, Chung J, van Huizen M, Teyra J, Miersch S, Luu GK, Ye W, Sharma N, Ganaie SS, Russell R, Chen C, Maynard M, Amarasinghe GK, Mark BL, Kikkert M, Sidhu SS (2022) Ubiquitin variants potently inhibit SARS-CoV-2 PLpro and viral replication via a novel site distal to the protease active site. PLOS Pathogens 18(12): e1011065. https://doi.org/10.1371/journal.ppat.1011065
- Wang C, Greene D, Xiao L, Qi R, Luo R (2018) Recent developments and applications of the MMPBSA method. Frontiers in Molecular Biosciences 4: 87. https://doi.org/10.3389/fmolb.2017.00087
- Wibowo ZK, Ramadhani AS, Rachman MA, Khaerunnisa S (2022) In Silico Analysis of Potential Nicotine Addiction Treatment by Cinnamomum verum Phytochemicals against nAChRα3 and nAChRα7. International Journal of Scientific Advances 3(1): 42–48. https://doi.org/10.51542/ijscia.v3i1.5
- Wurtzer S, Marechal V, Mouchel JM, Maday Y, Teyssou R, Richard E, Almayrac JL, Moulin L (2020) Evaluation of lockdown effect on SARS-CoV-2 dynamics through viral genome quantification in waste water, Greater Paris, France, 5 March to 23 April 2020. Eurosurveillance 25(50): 2000776. https://doi.org/10.2807/1560-7917.ES.2020.25.50.2000776
- Yapasert R, Khaw-On P, Banjerdpongchai R (2021) Coronavirus infection-associated cell death signaling and potential therapeutic targets. Molecules 26(24): 7459. https://doi.org/10.3390/molecules26247459
- Zhao G, Liu X, Wang S, Bai Z, Zhang S, Wang Y, Yu H, Xu X (2022) Hydrogen bonding penalty used for virtual screening to discover potent inhibitors for Papain-Like cysteine proteases of SARS-CoV-2. Chemical Biology and Drug Design 100(4): 502–514. https://doi.org/10.1111/cbdd.14115
- Zorofchian Moghadamtousi S, Abdul Kadir H, Hassandarvish P, Tajik H, Abubakar S, Zandi K (2014) A review on antibacterial, antiviral, and antifungal activity of curcumin. BioMed Research International 2014: 186864. https://doi.org/10.1155/2014/186864
- Zupin L, Fontana F, Clemente L, Borelli V, Ricci G, Ruscio M, Crovella S (2022) Optimization of Anti-SARS-CoV-2 Treatments Based on Curcumin, Used Alone or Employed as a Photosensitizer. Viruses 14(10): 2132. https://doi.org/10.3390/v14102132