Research Article |
|
Corresponding author: Ruswanto Ruswanto ( ruswanto@universitas-bth.ac.id ) Academic editor: Maya Georgieva
© 2023 Ruswanto Ruswanto, Richa Mardianingrum, Ade Dwi Septian, Arry Yanuar.
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Ruswanto R, Mardianingrum R, Septian AD, Yanuar A (2023) The design and virtual screening of thiourea derivatives as a Sirtuin-1 inhibitor. Pharmacia 70(4): 1335-1344. https://doi.org/10.3897/pharmacia.70.e108012
|
Abstract
The SIRT1 is overexpressed in a number of cancers. As a result, inhibiting SIRT1 may be used as a cancer treatment technique. Modification of 1-benzoyl-3-methylthiourea derivatives was carried out in this study by changing the aromatic side. The designed compounds (94) were subjected to in silico docking, pharmacokinetics, and preclinical testing. In the docking sample, the (4-decyl-N-(methylcarbamothioyl)benzamide (91), 2-(benzyloxy)-N-(methylcarbamo-thioyl)benzamide (93) and N-(methylcarbamothioyl)-2-naphthamide (94) were predicted to display better inhibition of SIRT1, so they were chosen for subsequent molecular dynamic studies. The compound 93 is proposed as a possible anticancer candidate that inhibits SIRT1 based on the screening results from molecular docking, pharmacokinetic predictions, and molecular dynamics.
Keywords
cancer, molecular docking, molecular dynamics, SIRT1, thiourea
Introduction
SIRT1 and SIRT2, which are part of the sirtuin family of enzymes, have been identified as class III histone deacetylases. Sirtuin was originally recognized as a yeast silent information regulator 2 (Sir2) enzyme. So far, researchers have discovered seven mammalian sirtuins (SIRT1 to SIRT7) that play a role in maintaining genomic stability, responding to stress, regulating lifespan, and promoting tumor growth (
Regarding sequence, SIRT1, which is closest to yeast Sir2, mediates the formation of heterochromatin by histone deacetylation. The significant role in chromatin modulation and epigenetic alteration of histone H1, H3, and H4 is attributed to the deacetylation of specific lysine residues by SIRT1. SIRT1 also has non-histone protein substrates, including p53, FOXO and Rb. By deacetylating these substrates, SIRT1 has been associated with various physiological functions (
It seems that SIRT1 and SIRT2 are involved in the progression of tumors. Elevated levels of SIRT1 have been detected in various cancer types, such as leukemia, lymphoma, skin cancer, breast cancer, liver cancer, stomach cancer, and colorectal cancer. The role of SIRT1 in tumorigenesis depends on the context, and studies examining whether SIRT1 functions as a tumor suppressor have produced inconsistent results. The anticancer effects of small-molecule inhibitors of SIRT1/2 aid the ability of SIRTs to promote tumor growth. Several small molecules have been discovered and suggested as potential treatments for cancer (
Thiourea is a chemical that is commonly utilized in drug discovery and development. In previous research, synthesis and activity tests were carried out on cancer cells with several derivatives of 1-benzoyl-3-methyl thiourea (
Materials and methods
Materials
The hardware utilized is a personal computer with the following specifications: Intel(R) Core(TM) i5-8265U CPU @ 1.60GHz (8 CPUs) × 8.00 GB Ram 64-Bit Windows 10 Operating System. The AutodockTools 1.5.6, MarvinSketch 21.17.0, Biovia Discovery Studio Visualizer, Amber 2016, and web-based applications such as PDB (Protein Data Bank), ProSA, SwissADME, PreADMET, and pkCSM were utilized.
SIRT1 structure retrieval and assessments
The SIRTUIN-1 enzyme’s three-dimensional structure (PDB ID 4I5I), which comes from Homo sapiens, was acquired from the protein database of the Structural Bioinformatics Research Collaborators (https:/www.rcsb.org/structure/4I5I). The crystal structure analysis of the catalytic domain of SIRT1 in conjunction with nicotinamide adenine dinucleotide (NAD+) and an indole compound (an analog of EX527) has revealed a new mechanism of histone deacetylase inhibition, with a resolution of about 2.5 angstroms.
The data was saved in PDB format, and the structural characteristics were evaluated. The stereochemical characteristics of the 3D structure were first explored using the Ramachandran plot. Additional properties of the 3D structure were studied, and their coherence was validated using the pdbsum and ProSA online interfaces.
The Ramachandran plot is a valuable tool for illustrating the phi (Φ) and psi (Ψ) dihedral angles of amino acid residues within a protein’s structure, identifying the protein structure’s allowed and disallowed conformations. The overall consistency and viability of the protein structure are calculated by the ProSA (https://prosa.services.came.sbg.ac.at/prosa.php) server. The performance outcomes should fall within the scope of experimentally tested protein structures. The ProSA score determines the Z-score. It demonstrates the protein structure’s general coherence and the fold conformations’ dependability (
Macromolecule and ligand preparation
The Sirtuin-1 structure was obtained from the PDB with a resolution of 2.5 Å, identified by the PDB ID 4I5I (
The compound design was carried out from the development of the 1-benzoyl-3-methylthiourea, which had previously been synthesized and tested for its activity on several cancer cells. In this study, the compound was modified by replacing the different groups in the positions on the aromatic ring to become 94 derivative compounds (as in Table
Docking results of the three best compounds and the native ligand (EX527).
| Compound code | Structure | Binding affinity (kcal/mol) | Inhibition constant |
|---|---|---|---|
| EX527 |
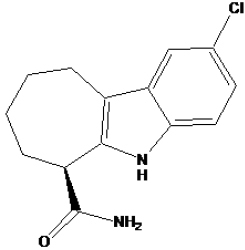
|
-9.71 | 73.09 nM |
| 91 |

|
-9.30 | 153.57 nM |
| 93 |
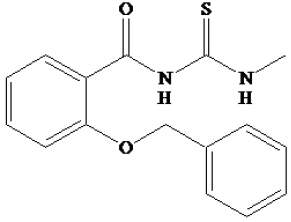
|
-8.76 | 381.19 nM |
| 94 |
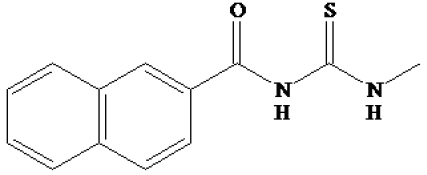
|
-8.31 | 813.74 nM |
The ninety-four of 1-benzoyl-3-methylthiourea derivatives were generated and illustrated in their 2-dimensional form. Subsequently, Marvin Sketch 5.2 software was utilized to conduct geometry optimization. Initially, the ligand was protonated at pH 7.4 to correspond with the pH of human blood in the body. The conformation was assessed to determine the molecular orientation that would be the most stable while interacting with the enzyme’s active site. To perform the docking process, the file was saved in two formats, .mrv and .pdb (
In silico pharmacology analysis and preclinical trials
Identifying different pharmacological characteristics and bioactivities, a pharmacology study and preclinical experiment were undertaken through web server applications such as SwissADME (http://www.swissadme.ch/index.php), http://pgp.biozyne.com/ and pKCSM (http://biosig.unimelb.edu.au/pkcsm/). Furthermore, the PreADMET system was used to forecast the absorption, distribution, and toxicity profiles of 1-benzoyl-3-methylthiourea derivatives (
Synthetic accessibility prediction
The prediction of synthesis accessibility is crucial in determining the feasibility of producing a more optimized medication in a laboratory setting. The SwissADME web interface was employed to evaluate the lead compounds’ practicality.
Molecular docking
AutoDockTools 1.5.6 was used to prepare the protein and ligands for docking (The Scripps Research Institute, La Jolla). To prepare the protein for docking, polar hydrogens, and Kollman charges were introduced and saved in .pdbqt format (
Molecular dynamics simulation
MD simulations were done on 1-benzoyl-3-methylthiourea, which had the lowest binding energy. A single trajectory approach was used to calculate all binding energies using MMGBSA. During the MD simulations, the AMBER ff14SB force field was used for modeling the protein. During the simulation, a general AMBER force field (GAFF) was used for the ligand, and a box with dimensions of 10 × 10 × 10 Å was filled with TIP3P water (Salomon-ferrer et al. 2012). The topology files were generated once the charges of the ligands were balanced using restricted electrostatic potential (RESP). The process of minimization, heating, and equilibration was conducted using the Sander module of Amber 16. The RMSD and RMSF were also determined. The native ligands were utilized as references. The docking and dynamics simulations were performed on a supercomputer equipped with Intel socket LGA1151, PCIE 3.0, good capacitors + Intel i5 Quad cores (4 cores/4 threads; 3.4 GHz) and the latest Kaby Lake processor (
Results and discussion
SIRT1 structure retrieval and assessments
The sirtuin-1 protein with human origin’s 3D structure (4I5I) was obtained from the PDB. Sirtuin-1 has a complex structure involving nicotinamide adenine dinucleotide (NAD+), an indole (EX527 analog), and zinc ions. The sirtuin-1 protein structure is composed of 272 amino acids. The three-dimensional structural properties were tested for molecular interaction studies (Fig.


Fig.
The ProSA program uses C-alpha atoms in a protein structure to calculate Z-scores, which compare the similarity of the structure to known structures of similar size determined by crystallography or NMR. If a model’s Z-score is negative, the model has few or no errors. The ProSA web server generated a Z-score of -8.74 for the 4I5I model, which is within the allowed range for similar-sized X-ray and NMR studies. Based on the observations in Fig.

Docking method validation
Docking method validation was performed with AutoDock software. Docking validation was performed by redocking the natural ligand ((6S)-2-chloro-5,6,7,8,9,10-hexahydrocyclohepta[b]indole-6-carboxamide, 415) in the 4I5I.pdb crystallographic structure onto its binding site. The docking process utilized a grid box with dimensions x = 42.8139 Å, y = -21.9161 Å, and z = 18.4876 Å. The parameter used for validation was the RMSD, which measures two postures made by comparing the atomic locations of the experimental and docked structures. The method is considered satisfactory if the resulting RMSD value is 2 or lower. The redocking results revealed an RMSD of 0.37 Å with a binding affinity of -9.71 kcal/mol and an inhibition constant of 75.71 nM. Overlay of the redocking 415 structures with the ones in the crystallographic structure (415I.pdb) as shown in Fig.

Docking and visualization results
Molecular interaction studies were conducted on the compounds that target the sirtuin-1 protein. Using AutoDock’s molecular docking methods, the generated sirtuin-1 protein structure was docked with the specified chemicals and EX527 (
From the docking results of the 1-benzoyl-3-methylthiourea derivatives (In Suppl. material




Due to the low binding affinities and favorable intermolecular interactions of the three compounds in molecular docking analysis, these compounds may be potential candidates for Sirtuin 1 enzyme inhibition in cancer therapy. The compounds’ preclinical and pharmacological properties were then assessed.
Based on the findings in Fig.
Analysis of the 2D visualization of the docking results and the types of interactions comprised by the complex of compound 93-SIRT1 in Fig.
For the analysis of the docking results for the compound 94-SIRT1 complex shown in Fig.
While in the EX527-SIRT1 complex, as shown in Fig.
Figs
In silico preclinical trial and pharmacokinetic prediction
Pharmacokinetic properties and toxicity prediction were evaluated in the earliest preclinical in silico studies of potential inhibitors 91, 93, and 94. To predict the pharmacokinetic properties, preADMET (http://preadmet.bmdrc.org/), pkCSM (http://biosig.unimelb.edu.au/pkcsm/prediction) and swissadme (http://www.swissadme.ch/index.php) interfaces were used. PkCSM can predict the maximum recommended daily dose of a ligand for its development potential as an anticancer agent.
The maximum tolerated dose for both drugs was determined using pkCSM. Compound 91 demonstrated a significant tolerance level, with a recommended tolerable dose of -0.148 log mg/kg/day, while the doses for compound 93 and 94 were 0.329 and 0.092 log mg/kg/day, respectively. The conclusions were based on the highest prescribed beginning dose for phase 1 clinical trials based on 1222 human clinical trial experimental evidence.
Whether the compounds were substrates for P-glycoprotein (P-gp) was another significant factor that was investigated during the preclinical study. P-gp is an efflux transporter that pumps medicines and other substances out of cells and their substrates, explaining why some tumors resist cancer chemotherapy. P-gp has been identified as an important drug carrier, leading to resistance to anticancer medicines such as imatinib, lonafarnib, and taxanes (
Therefore, using the Biozyne web interface (http://pgp.biozyne.com/), we evaluated compounds 91, 93, and 94 for whether or not they are P-gp substrates. As shown in Fig.

The next step of the computational study examined the compounds’ absorption, distribution, and toxicity. The ADMET test was conducted using the preADMET program (Table
Data on the predicted adsorption, distribution and toxicity of the compounds.
| No | Compound | Pharmacokinetic prediction | Toxicity prediction | ||||
|---|---|---|---|---|---|---|---|
| Caco2 (nm/sec) | % HIA | % PPB | Ames test | Carcino mouse | Carcino rat | ||
| 1 | EX527 | 21.5866 | 91.615 | 87.21 | Mutagen | + | - |
| 2 | 91 | 45.3267 | 94.886 | 100 | Mutagen | - | - |
| 3 | 93 | 28.9385 | 94.857 | 87.952 | Mutagen | - | - |
| 4 | 94 | 21.6553 | 94.817 | 84.541 | Mutagen | - | - |
From Table
During preclinical research, measuring the hazardous potential of substances is critical. LD50 values were used to determine the acute toxicity of drugs by measuring their relative toxicity according to the standard measurement. The LD50 value of a substance refers to the concentration of the substance that causes death in 50% of the tested animals. The LD50 estimates for compounds 91, 93, and 94 were 3.029, 2.627, and 2.56 mol/kg, respectively, based on a rat-tested model of over 10,000 chemical compounds.
The chronic toxicity of the chemicals in rats has been evaluated through oral administration, to determine the minimum effective dose that induces adverse effects. We discovered that a high concentration of compounds 91 (0.828 log mg/day), 93 (1.325 log mg/day), and 94 (1.078 log mg/day) has no negative effects on rats. The clinical trial conducted using a bioinformatics method indicates that the tested compounds possess the necessary pharmacokinetic qualities for medicine use. Further in vitro and bioassays could be conducted to develop these compounds as effective anticancer drugs.
Synthetic accessibility
Following computer-assisted drug discovery and development, the next stage entails evaluating these compounds in vitro and in vivo bioactivity to identify their potential for development. To evaluate the synthetic feasibility and viability of the compounds, it is important to assess their synthetic accessibility and potential as lead compounds. The prediction is based on the screening and informative research of individual fragment contributions and the refining of structures from a database including millions of previously synthesized molecules. Therefore, it was possible to estimate the compounds’ synthetic feasibility through a de novo synthesis approach.
The software called SwissADME was utilized to forecast the synthetic feasibility and effectiveness of the compounds as potential anticancer agents. These techniques can determine the synthetic feasibility percentage by considering the starting materials’ complexity and the compounds’ structural or residue complexity (
Synthetic accessibility analysis of the designed compounds using swissADME.
| Compound code | Synthetic accessibility score | Lead likeliness |
|---|---|---|
| 91 | 2.58 | No |
| 93 | 2.281 | No |
| 94 | 1.55 | No |
The compound will be easy to synthesize if it has a synthetic accessibility value close to 1. The SwissADME analysis suggests that the designed compounds are expected to be easily synthesized, as they all have a score close to 1. In addition, we found that they do not have lead drug likeliness.
Molecular dynamics simulation
To evaluate the stability of the interactions that occur in molecular docking, the next step was to carry out MD simulation on the three best compounds (91, 93, and 94 and EX527) of about 94 1-benzoyl-3-methylthiourea derivatives. MD runs of 100,000 ps (100 ns) was carried out on the complexes of 91, 93, and 94 compounds with SIRT1. The RMSD of the heavy atoms of SIRT1 over 100 ns can be seen in Fig.

Fig.

The residual movement patterns were similar among all the complexes, as indicated by the RMSF outcomes. According to the analysis, the protein’s amino and carbon terminals displayed significant flexibility, whereas most residues exhibited tight flexibility. The amino acid residues that had the highest fluctuation were ARG274 and CYS374, while the amino acid residues of VAL258 and ILE437 had the lowest fluctuation. This suggests that the binding of the ligand did not cause a significant change in protein conformation.
The compound 93-SIRT1 complex trajectory was visualized to observe the ligand’s position and analyze its interaction. The visualization of their trajectory is shown in Fig.

From Fig.
MMGBSA calculations were performed to assess the binding affinity of each compound for SIRT1, and the results were presented in (Table
The MMGBSA energy was used to break down the calculated binding energies.
| Energy component | System | |||
|---|---|---|---|---|
| EX527-SIRT1 | Comp. 91-SIRT1 | Comp. 93-SIRT1 | Comp. 94-SIRT1 | |
| Vdw | -20.15±3.40 | -53.49±4.92 | -60.28±3.51 | -27.39±4.39 |
| EEL | 16.65±27.24 | 18.59±19.91 | -49.79±23.53 | 0.97±20.78 |
| EGB | -12.64±25.98 | -1.16±17.97 | 25.59±22.92 | 3.36±18.46 |
| ESUR | -2.08±0.32 | -6.39±0.54 | -5.14±0.16 | -3.17±0.45 |
| ∆Ggas (VdW+EEL) | -3.49±28.19 | -34.91±22.38 | -110.07±23.15 | -26.43±20.92 |
| ∆Gsolv (EGB + ESURF) | -14.72±25.89 | -7.55±17.68 | 20.45±22.92 | 0.18±18.49 |
| ∆GMMGBSA | -18.21±4.49 | -42.45±7.67 | -89.62±8.54 | -26.25±5.93 |
Compound 93 demonstrated a stronger intermolecular interaction with SIRT1 than other ligands, indicating that it formed more interactions with the receptor than compound 91, compound 94, and EX527. Van der Waals and electrostatic energies between compounds and receptors were calculated to measure their interactions. This was further supported by the observation that compound 93 had more surrounding residues and produced more hydrogen bonds (according to the hydrogen bond analysis) than the other ligands, indicating stronger interaction with SIRT1. However, the structure of compound 93 is larger than the others, leading to better van der Waals interactions. Most of the binding site residues were discovered to be hydrophobic amino acids. Furthermore, compound 93 has a reduced binding energy to the SIRT1 receptor -89.62±8.54 kcal/mol) than the others suggest that compound 93 prefers to bind SIRT1, so it could be recommended as a potential anticancer candidate drug to inhibit SIRT1.
Conclusions
Based on the results of virtual screening through docking, pharmacokinetics prediction, preclinical trial predictions, and molecular dynamics, compound 93 was the best potential anticancer candidate drug through the inhibition of SIRT1.
The positive findings of this work could serve as a foundation for future oncology research, specifically addressing SIRT1 inhibitors. Advanced clinical trials, exploring structural variations, mechanistic studies, and synergistic effects with other therapies are some of the prospective avenues and opportunities.
Conflict of interest
The authors do not have any competing interests to disclose.
Acknowledgements
This research was funded by the Penelitian Pasca Doktor Grant (Nomor: NKB-2964/UN2.RST/HKP.05.00/2020) from the Ministry of Research and Technology/BRIN Indonesia. We appreciate the facilities at STIKes Bakti Tunas Husada and Universitas Perjuangan Tasikmalaya.
References
- Adelin T (2013) Penambatan molekuler kurkumin dan analognya pada enzim siklooksigenase-2. Jurnal Medika Veterinaria 7(1): 30–34.
- Aromatic O, Carcinogens A (1972) Carcinogens as frameshift mutagens: metabolites and derivatives of 2-acetylaminofluorene and other aromatic amine carcinogens. PNAS 69(11): 3128–3132. https://doi.org/10.1073/pnas.69.11.3128
- De Beer TAP, Berka K, Thornton JM, Laskowski RA (2014) PDBsum additions. Nucleic Acids Research 42: 292–296. https://doi.org/10.1093/nar/gkt940
- Bosch-Presegué L, Vaquero A (2011) The dual role of sirtuins in cancer. Genes & Cancer 2: 648–662. https://doi.org/10.1177/1947601911417862
- Carafa V, Altucci L, Nebbioso A (2019) Dual tumor suppressor and tumor promoter action of sirtuins in determining malignant phenotype. Frontiers in Pharmacology 10: 1–14. https://doi.org/10.3389/fphar.2019.00038
- Chastagner P, Sudour H, Mriouah J, Barberi-Heyob M, Bernier-Chastagner V, Pinel S (2015) Preclinical studies of pegylated- and non-pegylated liposomal forms of doxorubicin as radiosensitizer on orthotopic high-grade glioma xenografts. Pharmaceutical Research 32: 158–166. https://doi.org/10.1007/s11095-014-1452-x
- Chi S, Ohue M, Gryniukov A, Borysko P et al. (2019) A prospective compound screening contest identified broader inhibitors for Sirtuin 1. Scientific Reports 9: 19585. https://doi.org/10.1038/s41598-019-55069-y
- Choi G, Lee J, Ji JY, Woo J, Kang NS, Cho SY, Kim HR, Ha JD, Han S-Y (2013) Discovery of a potent small molecule SIRT1/2 inhibitor with anticancer effects. International Journal of Oncology 43(4): 1205–1211. https://doi.org/10.3892/ijo.2013.2035
- Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug- likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 7: 42717 https://doi.org/10.1038/srep42717
- Erlina L, Yanuar A (2018) Molecular dynamics simulation of SIRT1 inhibitor from indonesian herbal database. Journal of Young Pharmacists 10(1): 3–6. https://doi.org/10.5530/jyp.2018.10.2
- Gertz M, Fischer F, Thi G, Nguyen T, Lakshminarasimhan M, Schutkowski M, Weyand M, Steegborn C (2013) Ex-527 inhibits Sirtuins by exploiting their unique NAD + -dependent deacetylation mechanism. PNAS 110(30): E2772–E2781. https://doi.org/10.1073/pnas.1303628110
- Hariono M, Nuwarda RF, Yusuf M, Rollando R, Jenie RI, Al-Najjar B, Julianus J, Putra KC, Nugroho ES, Wisnumurti YK, Dewa SP, Jati BW, Tiara R, Ramadani RD, Qodria L, Wahab HA (2020) Arylamide as potential selective inhibitor for matrix metalloproteinase 9 (MMP9): design, synthesis, biological evaluation, and molecular modeling. Journal of Chemical Information and Modeling 60: 349–359. https://doi.org/10.1021/acs.jcim.9b00630
- Koushik V, Alvala M, Saketh D, Viswanadha S, Sriram D, Yogeeswari P (2014) Structure-based drug design of small molecule SIRT1 modulators to treat cancer and metabolic disorders. Journal of Molecular Graphics and Modelling 52: 46–56. https://doi.org/10.1016/j.jmgm.2014.06.005
- Kozako T, Suzuki T, Yoshimitsu M, Arima N, Honda S, Soeda S (2014) Anticancer agents targeted to sirtuins. Molecules 19(12): 20295–20313. https://doi.org/10.3390/molecules191220295
- Lanevskij K, Dapkunas J, Juska L, Japertas P, Didziapetris R (2011) QSAR analysis of blood – brain distribution: the influence of plasma and brain tissue binding. Journal of Pharmaceutical Sciences 100(6): 2147–2160. https://doi.org/10.1002/jps.22442
- Lee HN, Jang HY, Kim HJ, Shin SA, Choo GS, Park YS, Kim SK, Jung JY (2016) Antitumor and apoptosis-inducing effects of α -mangostin extracted from the pericarp of the mangosteen fruit (Garcinia mangostana L .) in YD-15 tongue mucoepidermoid carcinoma cells. International Journal of Molecular Medicine 37(4): 939–948. https://doi.org/10.3892/ijmm.2016.2517
- Li H, Li H, Qu H, Zhao M, Yuan B, Cao M, Cui J (2015) Suramin inhibits cell proliferation in ovarian and cervical cancer by downregulating heparanase expression. Cancer Cell International 15: 1–11. https://doi.org/10.1186/s12935-015-0196-y
- Lilly E (2011) Sirtuin 1 (SIRT1): The Misunderstood HDAC. SLAS Discovery 16(10): 1153–1169. https://doi.org/10.1177/1087057111422103
- Ma W, Zhao X, Wang K, Liu J, Huang G (2018) Dichloroacetic acid (DCA) synergizes with the SIRT2 inhibitor Sirtinol and AGK2 to enhance anti-tumor efficacy in non-small cell lung cancer. Cancer Biology & Therapy 19: 835–846. https://doi.org/10.1080/15384047.2018.1480281
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser K, Simmerling C (2015) ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation 11(8): 3696–3713. https://doi.org/10.1021/acs.jctc.5b00255
- Mardianingrum R, Endah SRN, Suhardiana E, Ruswanto R, Siswandono S (2021) Docking and molecular dynamic study of isoniazid derivatives as anti-tuberculosis drug candidate. Chemical Data Collections 32: 100647. https://doi.org/10.1016/j.cdc.2021.100647
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Software News and Updates 30(16): 2785–2791. https://doi.org/10.1002/jcc.21256
- Muchtaridi M, Yusuf M, Syahidah HN, Subarnas A, Zamri A, Bryant SD, Langer T (2019) Cytotoxicity of chalcone of eugenia aquea burm F. leaves against T47D breast cancer cell lines and its prediction as an estrogen receptor antagonist based on pharmacophore-molecular dynamics simulation. Advances and Applications in Bioinformatics and Chemistry 12: 33–43. https://doi.org/10.2147/AABC.S217205
- Nebbioso A, Pereira R, Khanwalkar H, Matarese F, García-Rodríguez J, Miceli M, Logie C, Kedinger V, Ferrara F, Stunnenberg HG, de Lera AR, Gronemeyer H, Altucci L (2011) Death receptor pathway activation and increase of ROS production by the triple epigenetic inhibitor UVI5008. Molecular Cancer Therapeutics 10(12): 2394–2404. https://doi.org/10.1158/1535-7163.MCT-11-0525
- Pal S, Kumar V, Kundu B, Bhattacharya D, Preethy N, Reddy MP, Talukdar A (2019) Ligand-based pharmacophore modeling, virtual screening and molecular docking studies for discovery of potential topoisomerase I inhibitors. Computational and Structural Biotechnology Journal 17: 291–310. https://doi.org/10.1016/j.csbj.2019.02.006
- Park EY, Woo Y, Kim SJ, Kim DH, Lee EK, De U, Kim KS, Lee J, Jung JH, Ha K, Choi WS, Kim IS, Lee BM, Yoon S, Moon HR, Kim HS (2016) Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 cells via regulation of MDM2-p53 binding. International Journal of Biological Sciences 12(12): 1555–1567. https://doi.org/10.7150/ijbs.13833
- Prajapat R, Marwal A, Gaur RK (2014) Recognition of errors in the refinement and validation of three-dimensional structures of AC1 proteins of begomovirus strains by using ProSA-Web. Journal of Viruses 2014: 752656. https://doi.org/10.1155/2014/752656
- Qidwai T, Yadav DK, Khan F, Dhawan S, Bhakuni RS (2012) QSAR, docking and ADMET studies of artemisinin derivatives for antimalarial activity targeting plasmepsin II, a hemoglobin-degrading enzyme from P. falciparum. Current Pharmaceutical Design 18(37): 6133–6154. https://doi.org/10.2174/138161212803582397
- Qin Z, Kastrati I, Chandrasena REP, Liu H, Yao P, Petukhov PA, Bolton JL, Thatcher GRJ (2007) Benzothiophene selective estrogen receptor modulators with modulated oxidative activity and receptor affinity. Journal of Medicinal Chemistry 50(11): 2682–2692. https://doi.org/10.1021/jm070079j
- Ruswanto , Miftah AM, Tjahjono DH (2015) Synthesis and in vitro cytotoxicity of 1-benzoyl-3-methyl thiourea derivatives. Procedia Chemistry 17: 157–161. https://doi.org/10.1016/j.proche.2015.12.105
- Ruswanto , Siswandono , Richa M, Tita N, Tresna L (2017) Molecular docking of 1-benzoyl-3-methylthiourea as anti cancer candidate and its absorption, distribution, and toxicity prediction. Journal of Pharmaceutical Sciences and Research 9(5): 680–684.
- Ruswanto R, Mardianingrum R, Siswandono S, Kesuma D (2020) Reverse docking, molecular docking, absorption, distribution, and toxicity prediction of artemisinin as an anti-diabetic candidate. Molekul 15: 88–96. https://doi.org/10.20884/1.jm.2020.15.2.579
- Ruswanto R, Garna IM, Tuslinah L, Mardianingrum R, Lestari T, Nofianti T (2018) Kuersetin, penghambat uridin 5-monofosfat sintase sebagai kandidat anti-kanker. ALCHEMY Jurnal Penelitian Kimia 14(2): 236–252. https://doi.org/10.20961/alchemy.14.2.14396.236-254
- Salomon-ferrer R, Case DA, Walker RC (2013) An overview of the Amber biomolecular simulation package. WIREs Computational Molecular Science 3(2): 198–210. https://doi.org/10.1002/wcms.1121
- Schinkel AH, Smit JJM, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CAAM, der Valk MA, Robanus-Maandag EC, Riele HPJ, Berns AJM, Borst P (1994) Disruption of the mouse m & la P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77(4): 491–502. https://doi.org/10.1016/0092-8674(94)90212-7
- Shibata T, Yamagata T, Kawade A, Asakura S, Toritsuka N, Koyama N, Hakura A (2020) Evaluation of acetone as a solvent for the Ames test. Genes and Environment 42: 1–8. https://doi.org/10.1186/s41021-020-0143-6
- Sonnemann J, Kahl M, Siranjeevi PM, Blumrich A, Blümel L, Becker S, Wittig S, Winkler R, Krämer OH, Beck JF (2015) Reverse chemomodulatory effects of the SIRT1 activators resveratrol and SRT1720 in Ewing ’ s sarcoma cells: resveratrol suppresses and SRT1720 enhances etoposide – and vincristine – induced anticancer activity. Journal of Cancer Research and Clinical Oncology 142: 17–26. https://doi.org/10.1007/s00432-015-1994-2
- Sun Y, Zhou H, Zhu H, Leung S (2016) Ligand-based virtual screening and inductive learning for identification of SIRT1 inhibitors in natural products. Scientific Reports 6: 19312. https://doi.org/10.1038/srep19312
- Tan YJ, Lee YT, Yeong KY, Petersen SH, Kono K, Tan SC, Oon CE (2018) Anticancer activities of a benzimidazole compound through sirtuin inhibition in colorectal cancer. Future Medicinal Chemistry 10(17). https://doi.org/10.4155/fmc-2018-0052
- Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions Clean up structure. Protein Engineering, Design and Selection 8(2): 127–134. https://doi.org/10.1093/protein/8.2.127
- Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research 35(suppl_2): W407–W410. https://doi.org/10.1093/nar/gkm290
- Wössner N, Alhalabi Z, González J, Swyter S, Gan J, Sippl W, Jung M (2020) Sirtuin 1 inhibiting thiocyanates (S1th) – A new class of isotype selective inhibitors of NAD + dependent lysine deacetylases. Frontiers in Oncology 10: 1–15. https://doi.org/10.3389/fonc.2020.00657
- Zhang WEI, Yang R, Cieplak P, Luo RAY, Lee T, Caldwell J, Wang J, Kollman P (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase. Journal of Computational Chemistry 24(16): 1999–2012. https://doi.org/10.1002/jcc.10349
- Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, Ashok S, Russell M, Macewan I, Qian Y, Jamison JA, Luz JG (2013) The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD +) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. Journal of Medicinal Chemistry 56(3): 963–969. https://doi.org/10.1021/jm301431y
- Zhou AQ, Hern CSO, Regan L (2011) Revisiting the Ramachandran plot from a new angle. Protein Science 20(7): 1166–1171. https://doi.org/10.1002/pro.644
Supplementary material
The structure and docking results of the 1-benzoyl-3-methylthioureas derivatives
Data type: pdf