Research Article |
|
Corresponding author: Winarto Haryadi ( wnrt_haryadi@ugm.ac.id ) Academic editor: Irini Doytchinova
© 2023 Winarto Haryadi, Harno Dwi Pranowo.
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Haryadi W, Pranowo HD (2023) Molecular docking and dynamics analysis of halogenated imidazole chalcone as anticancer compounds. Pharmacia 70(2): 323-329. https://doi.org/10.3897/pharmacia.70.e101989
|
Abstract
Cancer is one of the three biggest causes of death in the world. The development of new drugs against this disease is a serious study that must be carried out in order to reduce mortality and extend the life span of sufferers. The aim of this research was to develop a new anti-cancer drug based on halogenated imidazole chalcones have been conducted. A 18 The halogenated imidazole chalcone compound was designed and performed molecular docking. Potential compounds of molecular docking are then carried out molecular dynamics. The results of molecular docking show that the potential chalcones based on bond affinity and specific interactions are chalcones B5, B6, C5, and C6. Analysis of the molecular dynamics result parameters are root mean standard deviation (RMSD) complex, root mean square fluctuation (RMSF), Radius of Gyration, Protein-ligand hydrogen bonding, and complex stability with generalized born surface area (GBSA) method, where the potential chalcone compounds are chalcones B5 and B6.
Keywords
imidazole-chalcone, anticancer, molecular docking, and molecular dynamics
Introduction
Cancer is a disease caused by abnormal and uncontrolled cell growth that can damage surrounding tissues. Cancer is a worldwide disease where there are 19.3 million new cancer cases followed by 10 million deaths. If not treated properly, it is estimated that there will be 28.4 million cancer cases annually by 2040 (
Chalcones are compounds with 2 aromatic rings connected by a α,β-unsaturated carbonyl skeleton and are compounds with extensive anti-cancer bioactivity (
In the development of new drug compounds it is very important to know the interactions that occur between an enzyme and an inhibitor because almost 95% of drug activity arises from these interactions (
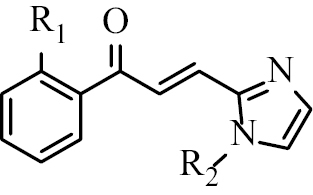
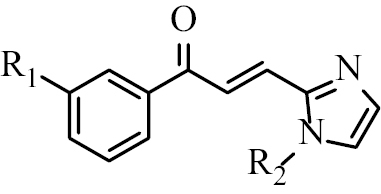
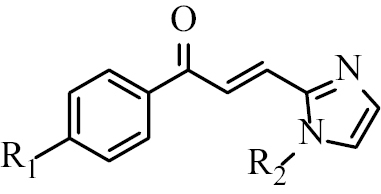
Based on this background, 18 halogenated chalcone compounds with imidazole skeletons (Table
| Compound Skeleton | Compound | |
|---|---|---|
|
Chalcone A1 | R1 = Cl R2 = H |
| Chalcone A2 | R1 = Br R2 = H | |
| Chalcone A3 | R1 = Cl R2 = CH3 | |
| Chalcone A4 | R1 = Br R2 = CH3 | |
| Chalcone A5 | R1 = Cl R2 = CH2-C6H5 | |
| Chalcone A6 | R1 = Br R2 = CH2-C6H5 | |
|
Chalcone B1 | R1 = Cl R2 = H |
| Chalcone B2 | R1 = Br R2 = H | |
| Chalcone B3 | R1 = Cl R2 = CH3 | |
| Chalcone B4 | R1 = Br R2 = CH3 | |
| Chalcone B5 | R1 = Cl R2 = CH2-C6H5 | |
| Chalcone B6 | R1 = Br R2 = CH2-C6H5 | |
|
Chalcone B1 | R1 = Cl R2 = H |
| Chalcone B2 | R1 = Br R2 = H | |
| Chalcone B3 | R1 = Cl R2 = CH3 | |
| Chalcone B4 | R1 = Br R2 = CH3 | |
| Chalcone B5 | R1 = Cl R2 = CH2-C6H5 | |
| Chalcone B6 | R1 = Br R2 = CH2-C6H5 | |
Materials and methods
Materials
Molecular docking procedure and dynamics performed using on a computer with specification Intel Xeon E5 2609 V4 with 16GB Ram, running open suse leap 15.3 operating system.
Molecular tethering
Eighteen chalcone compounds were 3D modeled with Marvin Sketch software and optimized for geometry with Gaussian 09W (Frisch et al. 2013) using the density functional theory (DFT) method with B3LIP hybrid function and set base 6–31G (d.p). Molecular tethering is performed with Auto Dock Vina 1.1.2 software (
Molecular dynamics
The three best compounds from molecular docking were studied for their stability on the active side. Molecular dynamics method with GROMACS software - The protein topology was created using pdb2gmx with the CHARMM-jul-2021 force field, The ligand topology was created using the CGenFF web. The system added water molecules and the system charge was neutralized by changing the Cl- anion (Tomanik et al. 2020). Furthermore, the system is minimized and continued with the equilibration of volume and temperature (NVT) and pressure and temperature (NPT) respectively for 100 and 250 ns. Molecular dynamics simulations were run within 35 ns with a system temperature of 300K and a pressure of 1 atm (
Result and discussion
Docking parameter validation
Re-tethering was performed on native ligand, namely erlotinib to validate the docking protocol The docking protocol used was center X, Y, Z = 23.907, 8.951, -0.436 with a grid box size of 20×20×20 Å followed by an exhaustiveness value of 64. The re-tethering results showed an RMSD value of 1.390 Å. The RMSD value of the re-tethering result indicates a value below 2 Å which means that the docking protocol used is valid and feasible for use in re-tethering the designed molecule (Fig.

Molecular docking
The results of molecular tethering showed that the chalcone compound substituted for the benzyl group in the imidazole skeleton, namely the chalcone (A5, A6, B5, B6, C5, and C6) had a lower bond affinity value than erlotinib. The observed bond affinity difference values of chalcones A5, A6, B5, B6, C5, and C6 are not too far apart compared to erlotinib. The not-so-distant difference in bond affinity is thermodynamically suspected that the designed ligands have the same ease of interacting with erlotinib on the active side of the EGFR protein.
In studying the results of molecular tethering, it is not only seen from the value of bond affinity but also seen from the interactions that occur on the active side. Specific interactions on the active side indicate that the designed compound interacts precisely with the active side and the presence of specific interactions also indicates the compound’s ability to close the interaction of active amino acid residues so that the catalytic process of a protein is inhibited. The active side and interaction that plays an important role in the inhibitory activity of the EGFR protein is the interaction of hydrogen bonds with the amino acid residue Met768 followed by the production of hydrophobic interactions Leu694, Val702, Ala719, Thr766 and Leu820 (

From the description of bond affinity and its interaction chalcone B5, B6, C5, and C6 it is very interesting to further study its stability in dynamic systems using molecular dynamics simulations (Table
| Compounds | Binding Affinity (kcal/mol) | Interactions | |
|---|---|---|---|
| Hydrogen Bonding | Hydrophobic interaction (Pi-pi stacking Pi-sigma/Pi-alkyl/alkyl) | ||
| Erlotinib (Native Ligand) | -7.9 | Met769 | Leu694, Val702, Ala719, Lys721, Leu768, Leu820 |
| Chalcone A1 | -7.0 | Lys721 | Leu694, Ala719, Leu820 |
| Chalcone A2 | -7.1 | Lys721 | Leu694, Ala719, Cys773 Leu820 |
| Chalcone A3 | -7.2 | - | Leu694, Val702, Ala719, Lys721, Thr766, Leu820 |
| Chalcone A4 | -7.2 | - | Leu694, Val702, Ala719, Lys721, Thr766, Leu820 |
| Chalcone A5 | -8.3 | Asp831 | Leu694, Val702, Ala719, Lys721, Thr766, Leu820 |
| Chalcone A6 | -8.2 | Asp831 | Leu694, Val702, Ala719, Lys721, Thr766, met769 Leu820 |
| Chalcone B1 | -7.1 | Lys721 | Leu694, Ala719, Leu820 |
| Chalcone B2 | -7.0 | - | Leu694, Val702, Ala719, Leu820 |
| Chalcone B3 | -7.4 | - | Leu694, Val702, Ala719, Leu820 |
| Chalcone B4 | -7.4 | - | Leu694, Val702, Ala719, Leu820 |
| Chalcone B5 | -8.1 | Met769 | Leu694, Val702, Ala719, Lys721 |
| Chalcone B6 | -8.2 | Met769 | Leu694, Val702, Ala719, Lys721 |
| Chalcone C1 | -6.8 | - | Leu694, Val702, Ala719, Leu768, Leu820 |
| Chalcone C2 | -6.8 | - | Leu694, Val702, Ala719, Leu820 |
| Chalcone C3 | -7.1 | Met769 | Leu694, Val702, Ala719, Leu820 |
| Chalcone C4 | -7.1 | - | Leu694, Val702, Ala719, Lys721, Thr766, Leu820 |
| Chalcone C5 | -8.1 | Met769 | Leu694, Val702, Ala719, Lys721 |
| Chalcone C6 | -8.1 | Met769 | Leu694, Val702, Ala719, Lys721 |
Evaluation of molecular dynamics results
The stability formed between proteins and ligands can be observed through changes in the root mean standard deviation (RMSD) value of protein and ligand complexes. RMSD is the value of the change in position of a structure to its initial position, and is directly related to the stability of the conformation of the structure. Measurement of the RMSD value in ligand atoms is important in assessing the entire complex of proteins and ligands and the smaller the change in RMSD value indicates the better the stability of the conformation (
The results of the RMSD analysis of potential chalcones from the molecular tethering results showed that the RMSD graph had a relatively similar pattern in which the RMSD value during the highest simulation was owned by the C6 chalcone with an average value of 0.47 nm, while the average RMSD values during the simulation for the B5 and B6 chalcones were 0.43 and 0.41 nm, respectively. The lowest average RMSD value during the simulation was owned by chalcone B5 with a value of 0.38. The average RMSD value of Chalcone B5, B6, and C5 is relatively close and lower than that of RMSD of C6 chalcone. If observed from the RMSD values, it can be seen that chalcone C5 as the most stable compound forms complexes with ligands. Sequentially During the simulation process, it was observed that before reaching 23 ns RMSD from each chalcone had different values, but the fluctuation patterns were relatively the same, the same relative fluctuation patterns occurred due to the similarity in structure of the chalcones. After 23 ns it was observed that the chalcone complexes B5, B6 and C6 showed a linear and overlapping graph indicating that the ligand protein complex had stabilized, but the RMSD value of the C6 chalcone complex showed fluctuations after 27 ns, in contrast to chalcones B5 and B6 which remained stable after 23 ns. At 25 ns RMSD the C5 chalcone complex showed a decrease in RMSD value but again followed the pattern of chalcones B5 and B6 after 32 ns (Fig.

The analysis continued by analyzing the root mean square fluctuation (RMSF) value of amino acid residues. Analysis of RMSF values shows the stability of the interactions that occur during the simulation process, which is calculated at each position of the amino acid residue and is characterized by the resulting fluctuations. From all the chalcones it is observed that the RMSF pattern formed is similar (Fig.


Analysis of radius of gyration shows the compactness of a protein ligand complex, the higher the radius of gyration value indicates that the reduced compactness of the complex formed (

| Compounds | Energy type (Kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ΔVDWAALS | ΔEEL | ΔEGB | ΔESURF | ΔGGAS | ΔGSOLV | ΔTOTAL | |
| Chalcone B5 | -32.93 | -23.21 | 36.61 | -4.06 | -56.14 | 32.55 | -23.59 |
| Chalcone B6 | -33.25 | -23.04 | 35.94 | -4.08 | -56.29 | 31.86 | -24.43 |
| Chalcone C5 | -35.13 | -17.13 | 33.68 | -4.53 | -52.26 | 29.15 | -23.11 |
| Chalcone C6 | -33.60 | -18.07 | 33.43 | -4.24 | -51.68 | 29.18 | -22.49 |
Hydrogen bonding during molecular dynamics simulation is related to the stability of the bond between proteins and ligands because hydrogen bonds are the largest contributors to stability when interacting with specific residues on the active side (
Complex stability energy calculations during simulations can be performed using the molecular mechanics generalized born surface area (MM-GBSA). This technique calculates the average of the energy changes that contribute to the process of complex formation (Wang el al., 2019). The results of the analysis of the energy of the stability of the complex are shown by Table
Reviewing the overall aspects of molecular dynamics parameters, information was obtained that the value of complex RMSD is not so much different, but the presence of a bromo group substituted at the meta position has a higher complex RMSD (chalcone C6). The position of the halogen substituent has no significant difference in the value of RMSF and Radius of Gyration, but the influence of the substituent position is seen in the hydrogen bond formed and the stability energy of the complex. The number of hydrogen bonds formed for chalcones whose halogens are meta-substituted is greater than that of the para-substituted. The stability energy of the complex also indicates the same thing where the stability energy of the complex is lower when the halogen is substituted in the meta position. Judging from these various parameters, the recommended compounds to be studied for activity in vitro are chalcone compounds B5 and B6.
Conclusions
The molecular tethering results of the 18 designed chalcone compounds show that the most potential compounds are chalcones B5, B6, C5, and C6 because they are able to form specific interactions with important residues of met769 and have similar hydrophobic interactions with native ligands. The results of molecular dynamics simulation show that the complex RMSD value is relatively the same where the meta-substituted chalcone has the highest RMSD is the C6 chalcone. The RMSF and radius of gyration values of the four chalcones did not have significant differences but there was an influence of substituent position seen in the hydrogen bonds formed and the stability energy of the complex. The compounds with the most hydrogen bonds and the most stable complex stability energy are those with halogen groups substituted in the meta position. Thus, the recommended compounds from various molecular dynamics parameter analysis are chalcones B5 and B6.
Acknowledgments
The author would like to thank Mr Joshua Eka and Mr Kasta Gurning who have helped during the research and the Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Gadjah Mada for founding this research through a research grant from the Department of Chemistry in 2022.
References
- Ahmadabadi MN, Rezaee E, Nematpour M, Karami L, Mokhtari S, Kobarfard F, Tabatabai SA (2022) Synthesis, Molecular Dynamics Simulation, and In-vitro Antitumor Activity of Quinazoline-2,4,6-triamine Derivatives as Novel EGFR Tyrosine Kinase Inhibitors. Iranian Journal of Pharmaceutical Research 21(1): 1–18. https://doi.org/10.5812/ijpr-133840
- Al-Anazi M, Khairuddean M, Al-Najjar BO, Alidmat MM, Kamal NNSNM, Muhamad M, Hariono M (2021) EGFR Inhibitors and apoptosis inducers: design, docking, synthesis, and anticancer activity of novel tri-chalcone derivatives. Systematic Reviews in Pharmacy 12(3): 809–820.
- Chaudhary N, Aparoy P (2017) Deciphering the mechanism behind the varied binding activities of COXIBs through molecular dynamic simulations, MM-PBSA binding energy calculations and per-residue energy decomposition studies. Journal of Biomolecular Structure and Dynamics 35(4): 868–882. https://doi.org/10.1080/07391102.2016.1165736
- Chouiter MI, Boulebd H, Pereira DM, Valentão P, Andrade PB, Belfaitah A, Silva AMS (2020) New chalcone-type compounds and 2-pyrazoline derivatives: Synthesis and caspase-dependent anticancer activity. Future Medicinal Chemistry 12(6): 493–509. https://doi.org/10.4155/fmc-2019-0342
- Do TH, Nguyen DM, Truong VD, Do THT, Le MT, Pham TQ, Thai KM, Tran TD (2016) Synthesis and selective cytotoxic activities on rhabdomyosarcoma and noncancerous cells of some heterocyclic chalcones. Molecules 21(3): 329. https://doi.org/10.3390/molecules21030329
- Fu Y, Zhao J, Chen Z (2018) Insights into the molecular mechanisms of protein-ligand interactions by molecular docking and molecular dynamics simulation: a case of oligopeptide binding protein. Computational and Mathematical Methods in Medicine 2018: 1–12. https://doi.org/10.1155/2018/3502514
- Karthikeyan C, Moorthy SHNN, Ramasamy S, Vanam U, Manivannan E, Karunagaran D, Trivedi P (2015) Recent patents on anti-cancer drug discovery advances in chalcones with anticancer activities. Recent Patents on Anti-Cancer Drug Discovery 10(1): 97–115. https://doi.org/10.2174/1574892809666140819153902
- Knapp B, Frantal S, Cibena M, Schreiner W, Bauer P (2011) Is an intuitive convergence definition of molecular dynamics simulations solely based on the root mean square deviation possible?. Journal of Computational Biology 18(8): 997–1005. https://doi.org/10.1089/cmb.2010.0237
- Lobanov MY, Bogatyreva NS, Galzitskaya OV (2008) Radius of gyration as an indicator of protein structure compactness. Molecular Biology 42(4): 623–628. https://doi.org/10.1134/S0026893308040195
- Oskuei SR, Mirzaei S, Jafari-Nik MR, Hadizadeh F, Eisvand F, Mosaffa F, Ghodsi R (2021) Design, synthesis and biological evaluation of novel imidazole-chalcone derivatives as potential anticancer agents and tubulin polymerization inhibitors. Bioorganic Chemistry 112(April): 104904. https://doi.org/10.1016/j.bioorg.2021.104904
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, Overington JP (2016) A comprehensive map of molecular drug targets. Nature Reviews Drug Discovery 16(1): 19–34. https://doi.org/10.1038/nrd.2016.230
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians 71(3): 209–249. https://doi.org/10.3322/caac.21660
- Tomanik L, Muchova E (2020) Solvation energies of ion with ensemble cluster-continuum approach. Physical Chemistry Chemical Physics 22: 22357–22368. https://doi.org/10.1039/D0CP02768E
- Trot O, Olson AJ (2009) Software news and update autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31(2): 455–461. https://doi.org/10.1002/jcc.21334
- Wang X, Zhang H, Chen X (2019) Drug resistance and combating drug resistance in cancer. Cancer Drug Resistance 2(2): 141–160. https://doi.org/10.20517/cdr.2019.10
- Yadav IS, Nandekar PP, Shrivastava S, Sangamwar A, Chaudhury A, Agarwal SM (2014) Ensemble docking and molecular dynamics identify knoevenagel curcumin derivatives with potent anti-EGFR activity. Gene 539(1): 82–90. https://doi.org/10.1016/j.gene.2014.01.056